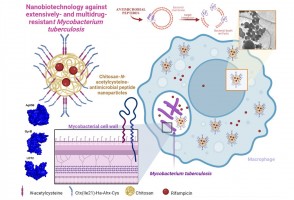
Cientistas da Unesp e da Universidade Estadual de Oklahoma verificaram em campo que liberação ideal de vespa que neutraliza o percevejo-marrom deve ser realizada de 30 em 30 metros

Estudo da USP, publicado na revista Environmental Research, analisou resultado das autópsias de 238 pessoas e dados epidemiológicos; perigo é maior para hipertensos
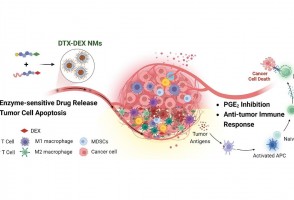
Em testes com animais, nanopartículas contendo substâncias já aprovadas para uso humano reduziram a inflamação no microambiente biológico em que cânceres desse tipo se instalam e vicejam, facilitando a ação do sistema imune

Estudantes e educadores da rede pública paulista têm até 3 de junho para se inscrever no edital, que aceita projetos de pesquisa em qualquer área do conhecimento envolvendo métodos da ciência para solucionar problemas
A missão, liderada pelo arqueólogo e antropólogo Walter Neves, objetiva entender como se deu o contato entre os Sapiens e os Neandertais. E por que estes desapareceram
Droga se acumula não só na água, mas em sedimentos e organismos marinhos e representa alto risco ecológico, apontou o professor da Unifesp Camilo Seabra durante a FAPESP Week Illinois
Trabalho com foco na transdisciplinaridade busca a participação dos diversos atores sociais para enfrentar as mudanças globais. Tema foi destaque em evento realizado este mês em São Luiz do Paraitinga
EPM-Unifesp
Inscrições até 30/04/2024
FMRP-USP
Inscrições até 30/04/2024
UFMG
Inscrições até 30/04/2024
EPM-Unifesp
Inscrições até 30/04/2024
RCGI/Poli-USP
Inscrições até 30/04/2024







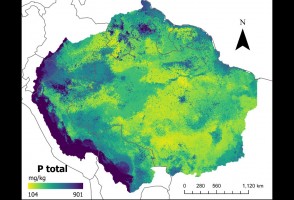


Último episódio da série de reportagens faz um balanço sobre a viagem de um grupo de pesquisadores do Museu de Zoologia da USP que, ao longo de duas semanas, percorreu os rios Negro, Preto e Jauaperi, nos Estados do Amazonas e de Roraima

Uma das artes de pesca utilizadas para coletar peixes-elétricos na Expedição DEGy Rio Negro foi empregada pela primeira vez em larga escala em água doce no projeto Calhamazon, que reuniu pesquisadores do Brasil e dos Estados Unidos entre 1993 e 1996

Baixa gravidade da lesão, atendimento médico rápido e cuidados adequados fizeram com que pesquisador pudesse voltar aos trabalhos no mesmo dia em que foi ferroado por peixe peçonhento. Na Amazônia, casos muitas vezes se agravam por carência de assistência especializada
02/03/2024 a 30/04/2024
30/03/2024 a 27/04/2024
06/04/2024 a 08/06/2024
09/04/2024 a 30/04/2024
22/04/2024 a 26/04/2024
25/04/2024 a 27/04/2024

Startup apoiada pelo PIPE-FAPESP está desenvolvendo helicóptero autônomo para pulverizar plantações em terreno íngreme

Startup apoiada pelo PIPE-FAPESP está desenvolvendo helicóptero autônomo para pulverizar plantações em terreno íngreme
Comunicar Ciência
Prazo: 22/01
Belmont Forum Climate, Environment, and Health
Prazo: Jan 2024
PIPE Start FAPESP-Sebrae: iniciando a jornada empreendedora de base tecnológica
Prazo: 18/03
Centros de Pesquisa em Inteligência Artificial Aplicada à Saúde
Prazo: 18/03
Fundação Nacional de Ciência da Suíça
Prazo: 22/03
Apoio a pesquisa em citricultura
Prazo: 31/03
Expedições Científicas Amazônia+10
Prazo: 29/04
EPM-Unifesp
Inscrições até 30/04/2024
FMRP-USP
Inscrições até 30/04/2024
UFMG
Inscrições até 30/04/2024
EPM-Unifesp
Inscrições até 30/04/2024
RCGI/Poli-USP
Inscrições até 30/04/2024
EPM-Unifesp
Incrições até 30/04/2024
FMRP-USP
Incrições até 30/04/2024
UFMG
Incrições até 30/04/2024
EPM-Unifesp
Incrições até 30/04/2024
RCGI/Poli-USP
Incrições até 30/04/2024