




Em sentido horário, ilustração da estrutura dímera da protease Mpro do SARS-CoV-2; simulação computadorizada da interação do inibidor N1 com Mpro; e representação bidimensional da molécula de N1, com destaque para as interações com os sítios ativos de Mpro (imagem: Journal of Biomolecular Structure and Dynamics)
Técnica foi aplicada por pesquisadores do Brasil, da Alemanha e da Finlândia no estudo de uma das principais proteínas que atuam no ciclo de reprodução do SARS-CoV-2, a Mpro
Técnica foi aplicada por pesquisadores do Brasil, da Alemanha e da Finlândia no estudo de uma das principais proteínas que atuam no ciclo de reprodução do SARS-CoV-2, a Mpro
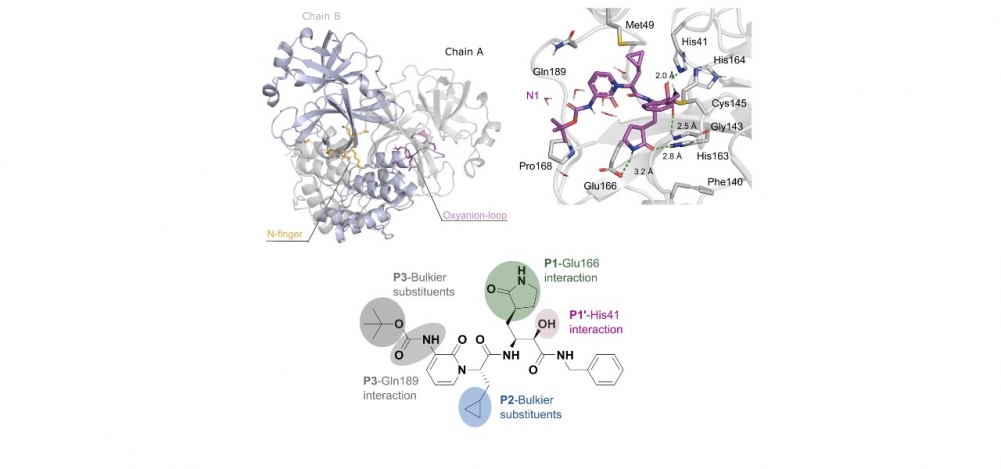
Em sentido horário, ilustração da estrutura dímera da protease Mpro do SARS-CoV-2; simulação computadorizada da interação do inibidor N1 com Mpro; e representação bidimensional da molécula de N1, com destaque para as interações com os sítios ativos de Mpro (imagem: Journal of Biomolecular Structure and Dynamics)
Janaína Simões | Agência FAPESP – Estudo conduzido por pesquisadores do Brasil, da Alemanha e da Finlândia propõe um novo padrão para simulações, utilizando técnicas computacionais, que promete acelerar a busca por compostos bioativos contra o vírus causador da COVID-19. O procedimento foi aplicado no estudo de uma das principais proteínas atuantes no ciclo reprodutor do SARS-CoV-2, que tem recebido grande atenção dos cientistas e da indústria farmacêutica por ser considerada um alvo para drogas antivirais. Os pesquisadores estimam que o método por eles desenvolvido possa reduzir o tempo gasto na fase inicial da pesquisa básica, que dura de dois a três anos, para menos de um ano.
Como explicam os autores, o SARS-CoV-2 apresenta uma camada externa de proteínas repletas do aminoácido cisteína, que precisa estar intacta e ativa para que o vírus mantenha sua atividade. A principal protease, enzima que quebra as ligações peptídicas entre aminoácidos e proteínas, é a Mpro (sigla derivada do termo em inglês main protease), responsável por clivar poliproteínas (cadeias proteicas) em proteínas menores. Estas, por sua vez, estão relacionadas à produção do RNA que codifica proteínas estruturais do patógeno, como a spike (que se liga ao receptor da célula humana viabilizando a infecção) e as que formam o envelope viral (camada mais externa que protege o material genético).
Pesquisadores e a indústria de fármacos olham a Mpro como alvo para o desenvolvimento de medicamentos contra a COVID-19 porque a possibilidade de influenciar ou bloquear essa clivagem representa a quebra de uma das etapas iniciais do ciclo de reprodução do vírus. Um dos caminhos é a sintetização de componentes químicos projetados para se ligar a pontos específicos da proteína, de forma a bloquear esse processo e inativá-la.
Para desenvolver novos medicamentos a indústria farmacêutica leva, em média, 20 anos. Um dos resultados do estudo publicado no Journal of Biomolecular Structure and Dynamics foi a construção de um padrão para simulações que pode reduzir para menos de um ano o tempo na busca por novos compostos bioativos contra o SARS-CoV-2. A procura pelo composto ativo é feita logo no início do desenvolvimento do medicamento, fase que normalmente leva de dois a três anos
“Para produzir as vacinas, foram usadas informações já relatadas do SARS-CoV-1, de quem o SARS-CoV-2 é uma evolução, acelerando a fase pré-clínica. Não podemos fazer o mesmo na pesquisa básica para desenvolver medicamentos porque não temos as informações de base necessárias para tal”, explica Glaucio Monteiro Ferreira, do Departamento de Análises Clínicas e Toxicológicas da Faculdade de Ciências Farmacêuticas da Universidade de São Paulo (FCF-USP) e autor principal do artigo. Ele conduziu parte da pesquisa no Departamento de Oncologia e Pneumonologia, Medicina Interna VIII do Hospital Universitário de Tübingen, na Alemanha, com apoio da FAPESP (16/12899-6 e 19/24112-9).
Outro fator que demanda mais tempo para a pesquisa e desenvolvimento de um novo fármaco são os estudos sobre a forma como ele será disponibilizado – se por via oral, como comprimido ou líquido, ou se na forma injetável, por exemplo. “Se for um medicamento para ser ingerido, precisamos garantir que ele passe por todas as barreiras do corpo humano para chegar aonde ele tem de fazer a ação. São detalhes que demandam mais tempo para desenvolver um fármaco se comparado a uma vacina”, completa.
Dados inéditos
Utilizando uma combinação de ferramentas, como a bioinformática e a biologia estrutural, Ferreira investigou a Mpro (também chamada de 3CLpro) – uma das proteínas virais que precisam ter o aminoácido cisteína como substrato ou “alimento” para cumprir sua função.
Diversos estudos anteriores mostram que existem proteínas que funcionam no estado monomérico, aquelas que têm apenas uma cadeia de proteína. “Mas sabíamos que o SARS-CoV-1 tem a cadeia de proteína duplicada, são dímeros. Isso complica a busca de um fármaco, pois precisamos encontrar um composto que consiga impedir que se forme a cadeia dupla”, conta.
Estudo anterior para o SARS-CoV-1 já tinha chegado a resultados promissores, usando inibidores covalentes ligados à cisteína – ou seja, um composto que se ligava de forma mais “forte” a esse aminoácido. A intenção desse estudo era encontrar um meio de impedir que a cisteína ficasse disponível para que a proteína se “alimentasse” dela, inibindo sua ação.
O SARS-CoV-2 também é um dímero. Para iniciar a busca por um composto que possa interferir na formação das duas cadeias, entram então as simulações. Usando a dinâmica molecular, método de simulação computacional que estuda o movimento físico dos átomos e moléculas, foi simulado um ataque do vírus em uma célula humana.
A equipe de cientistas integrada por Ferreira começou a fazer simulações em que a proteína tinha um e dois ligantes. Desse modo eles descobriram que as pesquisas focadas na análise das proteínas com uma cadeia utilizavam um experimento, a difração de raios X, que captava resultados depois que a proteína já tinha consumido a cisteína e se separado. Com isso, não conseguiam focar no processo de interesse, o bloqueio dessa fase na replicação da proteína.
Como forma de aplicar a simulação para o estudo do potencial de novos fármacos para combater o vírus da COVID-19, foram usados dois inibidores, o N1 e o N3, que afetam a ação da proteína Mpro. Os pesquisadores observaram que o primeiro é mais eficaz, não permitindo a doação de carga elétrica para a cisteína, ou seja, bloqueando a “alimentação” da proteína.
“Com as simulações que fizemos, chegamos mais rapidamente a esse inibidor, que pode realmente acabar com a enzima. Nossas simulações indicam o potencial desse composto ser um medicamento muito mais potente e estar muito mais próximo de virar um fármaco”, aponta. Coincidentemente, a Pfizer divulgou recentemente uma iniciativa de busca de medicamento para o SARS-CoV-2 que tem como alvo, justamente, um inibidor da proteína Mpro, o que ocorreu bem depois de iniciada a pesquisa de Ferreira.
O pesquisador brasileiro trabalhou em conjunto com Thales Kronenbergerb e Antti Poso, que atuam no Departamento de Oncologia e Pneumonologia, Medicina Interna VIII, Hospital Universitário de Tübingen, na Alemanha, e na Escola de Farmácia, Faculdade de Ciências da Saúde, Universidade da Finlândia Oriental, na Finlândia; Arun Kumar Tonduruc, do mesmo departamento no Universitário de Tübingen; Rosario Dominguez Crespo Hirata e Mario Hiroyuki Hirata, ambos do Departamento de Análises Clínicas e Toxicológicas da Faculdade de Ciências Farmacêuticas da USP. A pesquisa foi feita durante o pós-doutoramento de Ferreira, no Laboratório de Biologia Molecular Aplicado ao Diagnóstico e Farmacogenômica (LBMAD), sob supervisão do professor Hirata.
Todos assinam o artigo SARS-COV-2 Mpro conformational changes induced by covalently bound ligands, que pode ser lido em: www.tandfonline.com/doi/full/10.1080/07391102.2021.1970626.
A Agência FAPESP licencia notícias via Creative Commons (CC-BY-NC-ND) para que possam ser republicadas gratuitamente e de forma simples por outros veículos digitais ou impressos. A Agência FAPESP deve ser creditada como a fonte do conteúdo que está sendo republicado e o nome do repórter (quando houver) deve ser atribuído. O uso do botão HMTL abaixo permite o atendimento a essas normas, detalhadas na Política de Republicação Digital FAPESP.
