




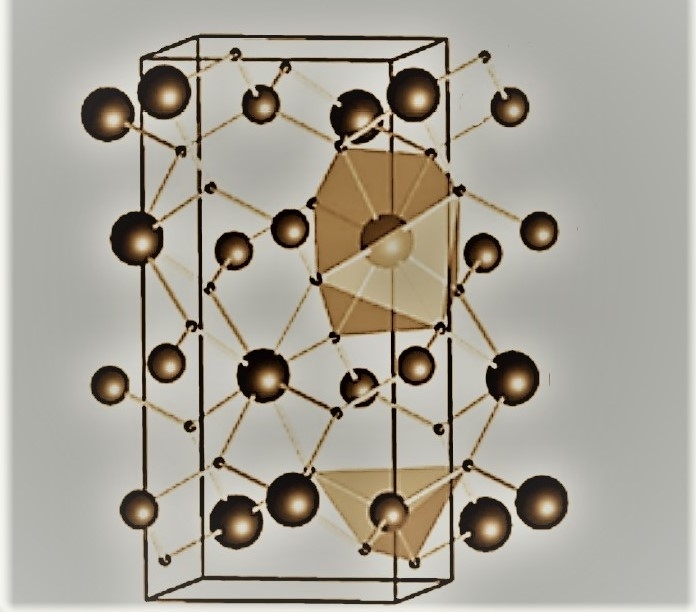
Un trabajo coordinado por un investigador brasileño facilita el análisis de materiales constituidos por una gran cantidad de átomos estructurados tridimensionalmente: un ejemplo es el molibdato de bario, con potencial aplicación tecnológica en luminiscencia y en la degradación de compuestos orgánicos (imagen: Ricardo Luis Tranquilin/ Unesp)
Un trabajo coordinado por un investigador brasileño facilita el análisis de materiales constituidos por una gran cantidad de átomos estructurados tridimensionalmente: un ejemplo es el molibdato de bario, con potencial aplicación tecnológica en luminiscencia y en la degradación de compuestos orgánicos
Un trabajo coordinado por un investigador brasileño facilita el análisis de materiales constituidos por una gran cantidad de átomos estructurados tridimensionalmente: un ejemplo es el molibdato de bario, con potencial aplicación tecnológica en luminiscencia y en la degradación de compuestos orgánicos
Un trabajo coordinado por un investigador brasileño facilita el análisis de materiales constituidos por una gran cantidad de átomos estructurados tridimensionalmente: un ejemplo es el molibdato de bario, con potencial aplicación tecnológica en luminiscencia y en la degradación de compuestos orgánicos (imagen: Ricardo Luis Tranquilin/ Unesp)
Por José Tadeu Arantes | Agência FAPESP – Puede resultar bastante complicado describir y explicar las propiedades electrónicas de ciertos materiales cristalinos constituidos por una estructura atómica ordenada tridimensionalmente, en razón de la gran cantidad de átomos implicados, cada uno de ellos a su vez con un gran número de electrones. Por eso se han desarrollado a tal fin modelos simplificados. Y esto fue lo que ocurrió con la Teoría del Funcional de la Densidad (DFT, del inglés Density Functional Theory), derivada de la mecánica cuántica y utilizada en física de sólidos y en química para dilucidar los sistemas de diversos cuerpos. En la DFT, las propiedades de los sistemas con muchos electrones se determinan mediante funcionales, es decir, mediante funciones de otra función que, en este caso, configuran la distribución espacial de la densidad electrónica.
La Teoría del Funcional de la Densidad, postulada en un artículo publicado en 1964 por el físico austroamericano Walter Kohn (1923-2016) y por el físico francoamericano Pierre Hohenberg (1934-2017), se empezó a aplicar ampliamente merced al desarrollo de los recursos computacionales. En 1998, Kohn recibió el Premio Nobel de Química en reconocimiento a su rol en la creación de la DFT.
Pero aún es necesario perfeccionar los métodos tendientes a obtener un modelado computacional más realista. Este fue el objetivo de un estudio que contó con el apoyo de la FAPESP y que se publicó en el periódico científico Computational Materials Science, que estuvo a cargo de Julio Ricardo Sambrano, coordinador del grupo de Modelado y Simulación Computacional de la Facultad de Ciencias de la Universidade Estadual Paulista (Unesp), con sede en la localidad de Bauru (estado de São Paulo). Este trabajo contó con la colaboración del profesor Elson Longo, director del Centro de Desarrollo de Materiales Funcionales (CDMF), un Centro de Investigación, Innovación y Difusión (CEPID) apoyado por la FAPESP y con sede en la Universidad Federal de São Carlos (UFSCar).
El material contemplado en este estudio fue el molibidato de bario (BaMoO4), elegido por su potencial aplicación tecnológica en luminiscencia y en la degradación de compuestos orgánicos. Actualmente se emplea el BaMoO4 para mejorar la adherencia de esmaltes y para remover el azufre de la nafta (un derivado del petróleo) en la producción de gases.
Agência FAPESP conversó con los profesores Sambrano y Longo acerca del trabajo que se ha publicado ahora.
Agência FAPESP – ¿Por qué es difícil describir y explicar las propiedades de ciertos materiales cristalinos?
Julio Ricardo Sambrano – Los materiales cristalinos poseen una estructura ordenada tridimensionalmente, con una enormidad de átomos, cada uno de los cuales está constituido por varios electrones. A cada uno de esos electrones puede representárselo con una función matemática. La situación ideal es la obtención de un modelo computacional con el cual se logre describir fielmente el sistema cristalino, es decir, concebir un modelo que contemple a todas las fuerzas que actúan en el sistema. Desafortunadamente, las limitaciones actuales de los modelos teóricos y la capacidad computacional disponible hacen que culminar esta tarea resulte imposible. Pero en un futuro cercano, y con el desarrollo de la computación cuántica, probablemente podrá modificarse esto. Un ejemplo: actualmente, con los cálculos de la mecánica cuántica, se efectúan una serie de aproximaciones: se empieza con la consideración de los núcleos atómicos en carácter fijo, cuando en la práctica se encuentran en constante movimiento. Las interacciones entre los electrones también son aproximadas para dos electrones.
Agência FAPESP – ¿Cómo ayuda la Teoría del Funcional de la Densidad (DFT) a resolver este problema?
Elson Longo – En los albores de la mecánica cuántica, pocos sistemas podían resolverse analíticamente, incluso mediante una solución numérica aproximada. Sin embargo, en 1927, el físico británico Llewellyn Thomas (1903-1992) y el físico italiano Enrico Fermi (1901-1954) propusieron un modelo probabilístico para el análisis de la densidad electrónica de los átomos. De esta idea surgió décadas más tarde la Teoría del Funcional de la Densidad, elaborada por Kohn y Hohenberg. Este modelo abrió dos puertas: la de la viabilidad de los cálculos de sistemas complejos como los cristales y la de la facilitación de la interpretación de los resultados. De este modo, y en forma coherente, se pueden interpretar las propiedades de sistemas tridimensionales, de superficies bidimensionales o incluso sistemas unidimensionales tales como nanotubos y nanohilos, y puede comparárselas con los resultados experimentales.
Agência FAPESP – ¿Por qué la DFT es una alternativa limitada?
Longo – No es solamente la DFT que es una alternativa que aún tiene sus limitaciones, sino que es también toda la mecánica cuántica la que se encuentra en constante avance en función del desarrollo de nuevas alternativas y metodologías. Esto se debe a la mayor disponibilidad computacional que, en este caso, hace posible el cálculo de sistemas cristalinos más complejos, el desarrollo de algoritmos más sofisticados y una mayor correlación entre los modelos y los resultados experimentales.
Agência FAPESP – ¿Cuáles son los nuevos métodos que pueden suministrar una representación más precisa de los materiales de interés?
Sambrano – El actual avance de la mecánica cuántica está pautándose según la interpretación de los datos teóricos en función de los resultados experimentales. El conocimiento de las energías de las distintas superficies de un cristal constituye una herramienta importante para producir modelos teóricos de las morfologías de un cristal a escala nanométrica. Estas morfologías pueden asociarse a los resultados experimentales obtenidos mediante microscopía electrónica de alta resolución. Y estos resultados pueden a su vez asociárselos con las propiedades de los cristales.
Agência FAPESP – ¿Cuáles son las posibles aplicaciones tecnológicas de una representación más precisa?
Longo – Para que exista generación de tecnología, el primer paso es el desarrollo del conocimiento. El refinamiento de los modelos teóricos hace posible lograr una mejor comprensión acerca de cómo experimenta una transformación una propiedad, en función de la perturbación de su densidad electrónica. De este modo, se puede conocer mejor un cristal y proponer nuevas investigaciones orientadas hacia una propiedad específica. Por ejemplo, mejorar el poder bactericida de un cristal y, simultáneamente, suministrar una mejor respuesta de sus propiedades fotoluminiscentes.
Agência FAPESP – ¿Cuál fue el programa computacional que se utilizó en este estudio?
Longo – Fue el programa CRYSTAL17, desarrollado por el grupo coordinado por el profesor Roberto Dovesi en la Università degli Studi di Torino, en Turín, Italia. Este programa permite ejecutar simulaciones a nivel DFT de cálculos de mecánica cuántica para estructuras periódicas como las de los cristales, y obtener las propiedades relacionadas para esos sistemas. El grupo de investigación que coordina el profesor Sambrano en la Unesp tiene una vasta experiencia en el área de simulaciones aplicadas a materiales y en el programa CRYSTAL. El profesor Sambrano pasó un largo período en Turín con el profesor Dovesi y mantiene una colaboración con los desarrolladores del programa. Esa asociación ya ha redundado en una serie de artículos publicados, cuyos temas están relacionados con las investigaciones del CDMF, que cuentan con el apoyo de la FAPESP.
Puede accederse al artículo intitulado Computational procedure to an accurate DFT simulation to solid state systems en el siguiente enlace: www.sciencedirect.com/science/article/abs/pii/S0927025619304756?via%3Dihub#!.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
