



Científicos de la Universidad de São Paulo constataron que la proteína spike del nuevo linaje viral interactúa mejor con el receptor celular humano ACE2, al cual se une el SARS-CoV-2 para viabilizar la infección (imagen: Jadson Santos / USP)
Científicos de la Universidad de São Paulo constataron que la proteína spike del nuevo linaje viral interactúa mejor con el receptor celular humano ACE2, al cual se une el SARS-CoV-2 para viabilizar la infección
Científicos de la Universidad de São Paulo constataron que la proteína spike del nuevo linaje viral interactúa mejor con el receptor celular humano ACE2, al cual se une el SARS-CoV-2 para viabilizar la infección
Científicos de la Universidad de São Paulo constataron que la proteína spike del nuevo linaje viral interactúa mejor con el receptor celular humano ACE2, al cual se une el SARS-CoV-2 para viabilizar la infección (imagen: Jadson Santos / USP)
Elton Alisson | Agência FAPESP – Investigadores de las facultades de Medicina (FMRP) y de Odontología (FORP) de la Universidad de São Paulo (USP), en su campus de la localidad de Ribeirão Preto, en Brasil, identificaron uno de los factores que vuelven más infecciosa a la nueva variante del coronavirus SARS-CoV-2: el linaje B.1.1.7, originario del Reino Unido y con dos casos confirmados en el país sudamericano por el Instituto Adolf Lutz, el laboratorio central de salud pública del estado de São Paulo.
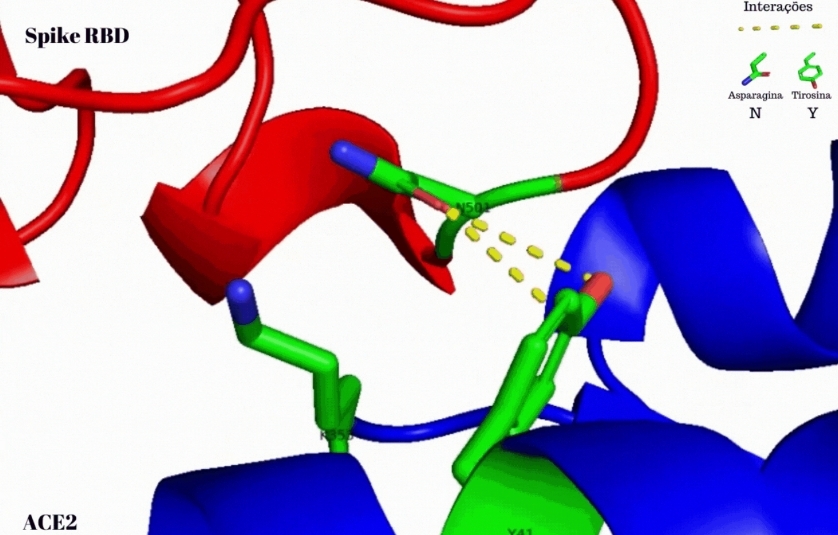
Mediante la aplicación de herramientas de bioinformática, los científicos constataron que la proteína spike de la nueva cepa viral –que forma la estructura en forma de corona que le da el nombre a esta familia de los coronavirus– establece una mayor fuerza de interacción molecular con el receptor ACE2, presente en la superficie de las células humanas y con el cual el SARS-CoV-2 se une para viabilizar la infección.
El incremento de la fuerza de interacción molecular del nuevo linaje es causado por una mutación ya detectada en el residuo de aminoácido 501 de la proteína spike del SARS-CoV-2, denominada N501Y, que dio origen a la nueva variante del virus, según aseveraron los investigadores.
Los resultados del trabajo, que contó con el apoyo de la FAPESP, salieron publicados en la plataforma bioRxiv, en un artículo aún sin revisión por pares.
“Observamos que la interacción entre la proteína spike de la nueva cepa del coronavirus con la mutación N501Y es mucho mayor que la que exhibía el primer linaje del virus aislado en Wuhan, en China”, le dice a Agência FAPESP Geraldo Aleixo Passos, docente de la FMRP y de la FORP-USP, y coordinador del proyecto. Otro autor del estudio, que estuvo a cargo de los análisis bionformáticos, es Jadson Santos, quien cursa un doctorado en la FMRP-USP bajo la dirección de tesis de Aleixo Passos.
Con el surgimiento del linaje B.1.1.7 en el Reino Unido, los investigadores plantearon la hipótesis de que la mutación N501Y presente en la proteína spike de la nueva variante, resultante del reemplazo de un aminoácido asparagina (N501) por uno tipo de tirosina (N501Y), podría constituir uno de los factores responsables de la alta contagiosidad del nuevo linaje del coronavirus.
Sucede que el N501 ya había sido identificado como un residuo de aminoácido crucial en la afinidad de unión de la proteína spike con el receptor ACE2 humano, y ello implicado en la infectividad del nuevo coronavirus. Asimismo, estudios anteriores también apuntaron que la mutación N501Y hallada en el linaje B.1.1.7 cubre uno de los seis residuos de aminoácidos de contacto o llaves dentro de la proteína spike.
“Existen otras mutaciones en el genoma de ese linaje que no analizamos. Nos enfocamos en la N501Y porque está implicada en la unión de la proteína spike con el ACE2”, explica Aleixo Passos.
Con el objetivo de poner a prueba la hipótesis de que la alta infectividad del linaje B.1.1.7 podría deberse a alteraciones en la fuerza de interacción entre la proteína spike mutante y el receptor ACE2, se utilizaron estructuras de la proteína spike del SARS-CoV-2 aislado en Wuhan y del linaje B.1.1.7, depositadas en un banco de datos de proteínas, el Protein Data Bank.
Mediante el empleo de un software de dominio público llamado PyMOL, fue posible visualizar la interacción entre el residuo de aminoácido 501 de la proteína spike del SARS-CoV-2 con el residuo Y41 de la proteína ACE2 humana y simular y analizar las interacciones resultantes de la mutación N501Y hallada en el linaje B.1.1.7 con el receptor celular.
“Este software permite visualizar imágenes de esas estructuras moleculares con una aproximación de 3.5 ángstroms de campo, mucho mayor que las imágenes generadas incluso con un ultramicroscopio”, compara Aleixo Passos.
Con otro software también de dominio público llamado PDBePISA, fue posible comparar la interacción de las proteínas spike del linaje silvestre del SARS-CoV-2 y del mutante con el receptor ACE2 humano.
Los resultados de los análisis mostraron que la mutación N501Y en la proteína spike de la nueva variante del coronavirus establece una mayor interacción con el receptor ACE2 en comparación con el linaje silvestre del virus. Las interacciones fueron predominantemente no covalentes –más débiles–, según afirmaron los investigadores.
“La sumatoria de varias uniones débiles entre la proteína spike mutante de la nueva variante del coronavirus con el receptor ACE2 humano resulta en interacciones moleculares más fuertes, que permiten que el virus ingrese más fácilmente a las células y active el sistema de replicación”, explica Aleixo Passos.
El estudio también reveló que la mutación N501Y provoca una alteración en el espaciamiento existente entre los residuos de aminoácidos de la proteína spike, lo que permite que la misma establezca aún más interacciones con el receptor ACE2.
“Juntas, estas alteraciones confirmaron la hipótesis que indica que la proteína spike de la cepa B.1.1.7 interactúa más fuertemente con el receptor ACE2”, afirma Aleixo Passos.
De acuerdo con el investigador, los resultados de este estudio, realizado mediante simulaciones computacionales in silico, permitirán orientar nuevos experimentos in vitro, destinados a evaluar en laboratorio la infectividad de la nueva variante del coronavirus en cultivos de células humanas.
Una evolución sorprendente
Según los investigadores, la rápida propagación del SARS-CoV-2 entre los humanos está impulsando su evolución molecular. Hasta ahora, el virus ha acumulado mutaciones a una tasa de hasta dos nucleótidos por mes, y aislados recientes exhiben al menos 20 alteraciones de nucleótidos en sus genomas en comparación con el linaje silvestre, aislado en enero de 2020. La mayor parte de las mutaciones se ubica en la proteína spike.
El linaje B.1.1.7, detectado al comienzo del mes de septiembre de 2020 y descrita en diciembre de 2020 por el COVID-19 Genomics UK Consortium, en el Reino Unido, y ya registrada en otros 17 países, Brasil inclusive, constituye un ejemplo, entre varios otros, de la rápida evolución molecular del nuevo coronavirus. Con todo, sorprendió a los científicos por acumular 17 mutaciones, de las cuales ocho se ubican en el gen que codifica a la proteína spike en la superficie del virus.
“Esta nueva cepa acumula muchas mutaciones. En otros linajes virales se observa una cantidad menor”, compara Aleixo Passos.
Como la descripción de esta nueva variante es reciente, aún no es posible evaluar con mayor minuciosidad el fenotipo, es decir, si es más o menos patogénica, según explica el investigador.
Puede accederse a la lectura del artículo intitulado The high infectivity of SARS-CoV-2 B.1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation (DOI: 10.1101/2020.12.29.424708), de Jadson C. Santos y Geraldo Aleixo Passos, en la plataforma bioRxiv, en el siguiente enlace: www.biorxiv.org/content/10.1101/2020.12.29.424708v1.full.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
