




Por meio de reprogramação celular, pesquisadores da USP descobriram que mutação genética está associada a formação anormal e morte prematura de neurônios. Na imagem da esquerda é possível observar neurônios derivados de células de um paciente com deficiência intelectual. À direita, neurônios derivados de células de um parente não afetado. (imagem: Thalita Figueiredo/CEGH-CEL)
Indivíduos portadores de uma condição rara apresentam mutação em um único gene que inibe ação de enzima importante para a formação de neurônios. Investigação foi realizada no Centro de Pesquisa sobre o Genoma Humano e Células-Tronco – um CEPID da FAPESP na USP com a colaboração do Salk Institute (EUA)
Indivíduos portadores de uma condição rara apresentam mutação em um único gene que inibe ação de enzima importante para a formação de neurônios. Investigação foi realizada no Centro de Pesquisa sobre o Genoma Humano e Células-Tronco – um CEPID da FAPESP na USP com a colaboração do Salk Institute (EUA)
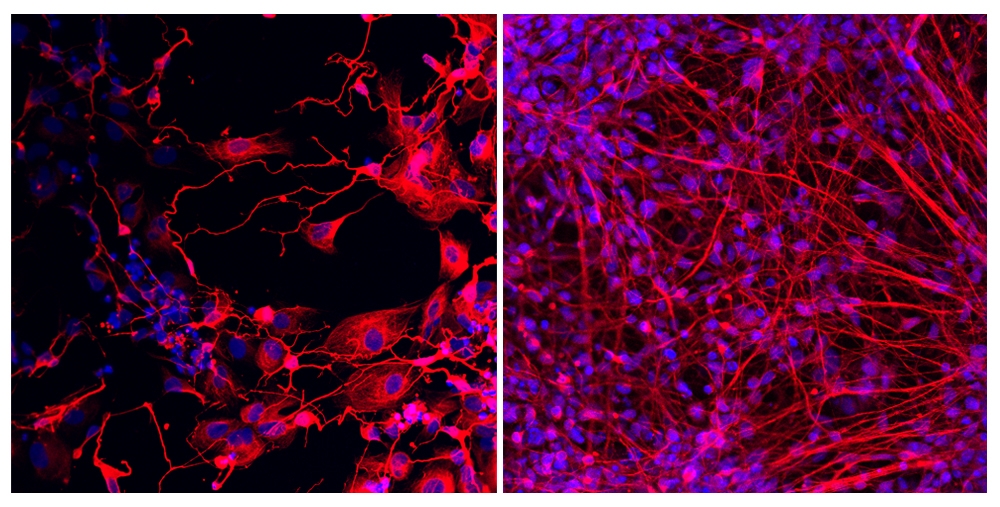
Por meio de reprogramação celular, pesquisadores da USP descobriram que mutação genética está associada a formação anormal e morte prematura de neurônios. Na imagem da esquerda é possível observar neurônios derivados de células de um paciente com deficiência intelectual. À direita, neurônios derivados de células de um parente não afetado. (imagem: Thalita Figueiredo/CEGH-CEL)
Maria Fernanda Ziegler | Agência FAPESP – Ao investigar casos de uma doença rara em Brejo dos Santos, uma cidadezinha isolada no sertão da Paraíba, pesquisadores da Universidade de São Paulo (USP) descobriram uma das causas para a deficiência intelectual grave. A descoberta, publicada na revista Molecular Psychiatry, amplia o entendimento sobre o desenvolvimento neurológico e abre caminho para novos estudos sobre doenças neurológicas e o desenvolvimento de novas drogas.
Na cidade, onde é relativamente comum o casamento consanguíneo, há alta incidência de uma doença rara. Numa só família, por exemplo, quatro dos dez filhos nasceram com a doença. Além de deficiência intelectual grave, eles também apresentam quadros psiquiátricos como alucinação, mania de perseguição e comportamento repetitivo.
“É uma doença rara que era desconhecida até então e o estudo pode também trazer pistas valiosas sobre os mecanismos envolvidos na formação de neurônios. É importante investigar e realizar estudos com essas populações, não apenas para descobrir e possivelmente tratar novas doenças. É a partir desses resultados que ampliamos conhecimento e podemos criar, no futuro, modelos capazes de elucidar as causas genéticas das mais diferentes doenças neurológicas”, explica Mayana Zatz, coordenadora do Centro de Pesquisa sobre o Genoma Humano e Células-Tronco (CEGH-CEL) e coautora do artigo.
O estudo, que teve a primeira publicação em 2012, a partir de uma pesquisa de campo no interior da Paraíba, teve a parte de reprogramação celular realizada no CEGH-CEL – um Centro de Pesquisa, Inovação e Difusão (CEPID) financiado pela FAPESP na USP. A pesquisa também integra um Projeto Temático, apoiado pela FAPESP, que estuda envelhecimento e doenças genéticas.
A investigação genética mostrou que a doença – identificada como deficiência intelectual autossômica recessiva – era causada pela mutação de um único gene (IMPA1). Mais recentemente, os pesquisadores buscaram entender os efeitos dessa mutação e descobriram que a alteração genética está associada à ausência de uma enzima (inositol), causando assim a formação anormal de neurônios.
“A deficiência intelectual pode ser causada por vários fatores genéticos e outros tantos ambientais. Na questão genética o mais comum é que seja causado por mutações em mais de um gene. Não é o caso dos pacientes que estudamos. Para manifestar a doença, a criança precisa receber um alelo com a mutação de sua mãe e outro de seu pai. O risco de isso acontecer é de 25% em cada gestação”, explica Thalita Figueiredo, primeira autora do estudo fruto da Bolsa de Pós-Doutorado e Bolsa Estágio de Pesquisa no Exterior, realizada no Salk Institute For Biological Studies (Estados Unidos), ambas apoiadas pela FAPESP.
Para descobrir que a mutação no gene IMPA1 desencadeava a doença, os pesquisadores coletaram amostras de sangue de indivíduos da família (todos adultos) com e sem a doença e realizaram o sequenciamento genético completo do exoma – parte do genoma onde ficam os genes codificadores de proteína e, portanto, onde há mais chance de ocorrerem mutações causadoras de doenças.
“Embora tenha sido a primeira vez que o gene foi relacionado com a deficiência intelectual, o IMPA1 é um gene muito estudado. Ele é responsável por produzir uma enzima que está envolvida no metabolismo do inositol, popularmente conhecido como um tipo de vitamina B e fundamental para várias funções celulares. Outro aspecto importante dessa enzima é que ela é inibida pelo lítio, principal medicamento utilizado como estabilizador de humor em pacientes com transtorno bipolar”, diz.
Uma nova etapa do estudo foi realizada para o mecanismo que desencadeia a síndrome. Por meio de técnicas de reprogramação celular, os pesquisadores transformaram células de sangue e de pele de pacientes com a forma grave de deficiência intelectual em células-tronco pluripotentes, para então convertê-las em neurônios do hipocampo e em astrócitos.
“Com esse estudo conseguimos entender a importância dessa enzima na neurogênese. A mutação está associada a uma formação anormal dos neurônios. Pacientes com deficiência dessa enzima não conseguem produzir neurônios de forma eficaz. Ocorre a morte prematura dos neurônios e eles não conseguem se replicar durante o desenvolvimento”, diz Figueiredo.
O hipocampo é a região do cérebro que está envolvida com a memória, comportamento. “Com a reprogramação celular, foi possível investigar em laboratório os efeitos da mutação genética na formação dos neurônios. É possível acompanhar desde o início com as células-tronco, depois com uma estrutura parecida com o tubo neural até se tornar um neurônio maduro, ou seja, na mesma fase que os pacientes estão, pois são todos adultos”, diz.
Figueiredo destaca que essa mutação é letal quando reproduzida em modelo de experimentação animal. “A prole de camundongos não vinga e morre antes de completar o desenvolvimento intrauterino. Por isso a importância de ter um modelo experimental por reprogramação genética. Sem essa técnica não seria possível conduzir o estudo e entender os mecanismos que causam esse tipo de deficiência intelectual em humanos”, diz Figueiredo.
No estudo em laboratório, os pesquisadores ainda analisaram a suplementação de inositol na cultura celular obtida dos pacientes. “Houve uma melhora muito grande na formação de novos neurônios. Sem o inositol, surgiram apenas 20% dos neurônios esperados em condições normais, já com a suplementação houve um aumento considerável, atingindo 80% da quantidade de neurônios”, diz.
De acordo com Figueiredo, o estudo também possibilita um provável tratamento para a doença rara. “É claro que são necessários mais testes que confirmem isso, mas seria possível fazer a suplementação de inositol um pouco antes de a mulher ficar grávida para que não haja um problema tão grave na neurogênese. No caso dos indivíduos adultos com a doença rara, isso não deverá ter efeito, pois já são células maduras”, diz.
O artigo Inositol monophosphatase 1 (IMPA1) mutation in intellectual disability patients impairs neurogenesis but not gliogenesis (doi: 10.1038/s41380-020-00862-9), de Thalita Figueiredo, Ana P. D. Mendes, Danielle P. Moreira, Ernesto Goulart, Danyllo Oliveira, Gerson S. Kobayashi, Shani Stern, Fernando Kok, Maria C. Marchetto, Renata Santos, Fred H. Gage e Mayana Zatz, pode ser lido em www.nature.com/articles/s41380-020-00862-9.
A Agência FAPESP licencia notícias via Creative Commons (CC-BY-NC-ND) para que possam ser republicadas gratuitamente e de forma simples por outros veículos digitais ou impressos. A Agência FAPESP deve ser creditada como a fonte do conteúdo que está sendo republicado e o nome do repórter (quando houver) deve ser atribuído. O uso do botão HMTL abaixo permite o atendimento a essas normas, detalhadas na Política de Republicação Digital FAPESP.
