




Com uma abordagem de biologia de sistemas, pesquisadores brasileiros identificaram diversos genes que poderão ser explorados como alvos terapêuticos e como biomarcadores de predisposição à artralgia crônica (imagem: PLOS Pathogens e kjpargeter/Freepik)
Com uma abordagem de biologia de sistemas, pesquisadores brasileiros identificaram diversos genes que poderão ser explorados como alvos terapêuticos e como biomarcadores de predisposição à artralgia crônica
Com uma abordagem de biologia de sistemas, pesquisadores brasileiros identificaram diversos genes que poderão ser explorados como alvos terapêuticos e como biomarcadores de predisposição à artralgia crônica
Com uma abordagem de biologia de sistemas, pesquisadores brasileiros identificaram diversos genes que poderão ser explorados como alvos terapêuticos e como biomarcadores de predisposição à artralgia crônica (imagem: PLOS Pathogens e kjpargeter/Freepik)
Karina Toledo | Agência FAPESP – Ferramentas computacionais aplicadas à biologia estão revolucionando o modo de estudar o que acontece no interior das células durante uma infecção, ajudando a desvendar o mecanismo de doenças e a encontrar potenciais alvos terapêuticos.
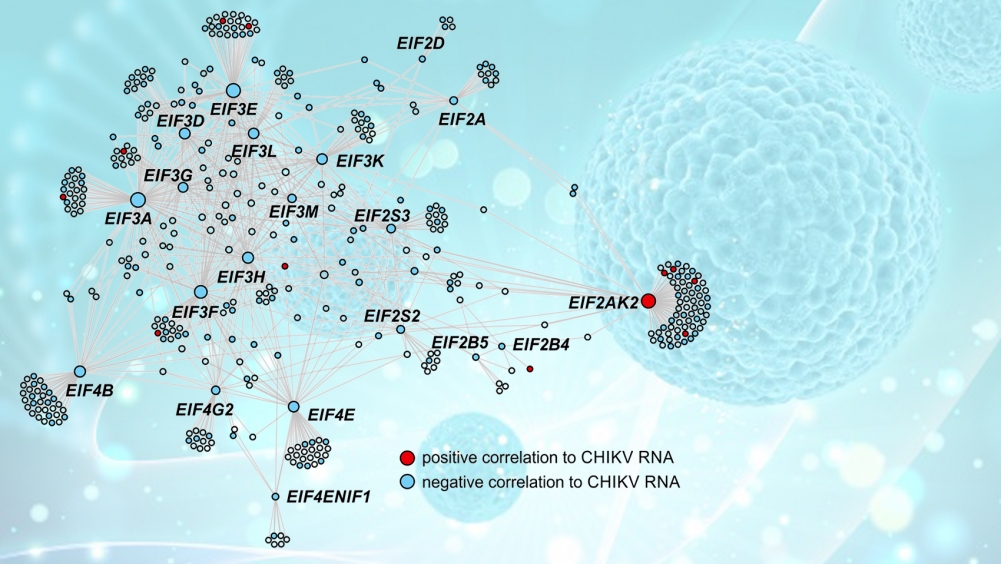
Este é o caso de um trabalho publicado recentemente na revista PLOS Pathogens, no qual pesquisadores brasileiros analisaram células sanguíneas de pacientes infectados pelo vírus chikungunya. Com auxílio de técnicas de análise de redes complexas, inteligência artificial e aprendizado de máquina, o grupo identificou a assinatura gênica da doença, ou seja, um conjunto de genes cuja expressão é alterada pela interação com o patógeno. Em seguida, o papel que os genes envolvidos desempenham nas células foi mapeado, bem como sua importância no combate ao vírus.
A pesquisa teve apoio da FAPESP e foi coordenada por Helder Nakaya, professor da Faculdade de Ciências Farmacêuticas (FCF) da Universidade de São Paulo (USP). Participaram colaboradores do Instituto de Ciências Biomédicas (ICB) da USP, da Faculdade de Medicina de Ribeirão Preto (FMRP-USP), do Instituto Butantan e do Laboratório Central de Saúde Pública de Sergipe, entre outros parceiros.
“Identificamos também um conjunto de genes capaz de indicar, ainda na fase aguda, se o paciente tende a evoluir para um quadro de artralgia crônica [inflamação nas articulações], relativamente comum em infectados por chikungunya. No entanto, esse achado ainda precisa ser confirmado por estudos futuros, feitos com uma quantidade maior de amostras”, disse Nakaya à Agência FAPESP.
O artigo traz resultados de análises feitas com amostras sanguíneas de 39 sergipanos infectados durante a epidemia de 2016. Os dados foram comparados com os de 20 controles – pessoas não infectadas e oriundas da mesma região dos pacientes estudados.
O primeiro passo foi analisar o transcritoma das amostras, ou seja, todas as moléculas de RNA mensageiro (que codificam proteínas) e também os RNAs não codificadores (que não dão origem a proteínas, mas têm ação reguladora no genoma) expressos nas células que compõem o sangue, como hemácias, leucócitos e plaquetas. Ao quantificar os transcritos nas amostras, os pesquisadores puderam medir o nível de atividade de 20 mil genes e avaliar, em comparação com os controles, quais ficavam com a expressão aumentada ou diminuída durante a infecção.
“Focamos nos genes codificadores de proteínas [aqueles que expressam os RNAs mensageiros], pois estes têm um papel mais fácil de ser interpretado. É relativamente simples saber se codificam um receptor celular ou um fator de transcrição, por exemplo. Conseguimos, assim, entender melhor a patogênese do chikungunya, isto é, como o vírus afeta as células e quais sistemas de defesa são ativados em resposta”, contou Nakaya.
Essa análise revelou, por exemplo, o mecanismo pelo qual as células imunes desencadeiam o processo inflamatório para eliminar o vírus. De modo geral, o conjunto de proteínas responsável por montar essa resposta de defesa é conhecido como inflamassoma. Trata-se de uma maquinaria celular que pode ser comandada por diferentes proteínas e resultar na produção de diferentes moléculas pró-inflamatórias. No caso da infecção pelo chikungunya, observou-se que a mediação é feita pela enzima caspase-1.
O achado foi validado por meio de experimentos com camundongos realizados em parceria com o pesquisador Dario Zamboni, da FMRP-USP. Os pesquisadores – ambos ligados ao Centro de Pesquisa em Doenças Inflamatórias (CRID), um CEPID da FAPESP sediado na USP de Ribeirão Preto – observaram que em animais geneticamente modificados para não expressar caspase-1 a infecção pelo chikungunya não induz a liberação da molécula pró-inflamatória chamada interleucina-1-beta (IL-1β) – ao contrário do que ocorre nos animais sem a alteração genética.
Similares, porém distintas
Após identificar a assinatura gênica da infecção por chikungunya – que envolve milhares de genes com expressão alterada na doença –, o grupo comparou os resultados com os obtidos em amostras de pacientes infectados pelo vírus da dengue.
“Notamos que ambas as assinaturas guardam certa similaridade, mas alguns genes são específicos para chikungunya. E são esses que poderão ser explorados em pesquisas voltadas ao desenvolvimento de fármacos”, disse Nakaya.
Em outra análise, os pesquisadores compararam o perfil de expressão gênica dos infectados pelo chikungunya com o de pessoas que sofrem de artrite reumatoide, doença autoimune caracterizada por inflamação crônica nas articulações.
“Nesse caso, o objetivo era descobrir a diferença entre a artrite causada pelo vírus, e a autoimune e identificar os genes específicos da infecção viral”, contou Nakaya.
A análise combinada das três assinaturas gênicas revelou 949 genes envolvidos apenas na artrite reumatoide, 632 apenas em dengue e 302 exclusivos de chikungunya. Sete genes apareceram nas três condições simultaneamente: OAS1, C1QB, ANKRD22, IRF7, CXCL10, IFI6 e IFIT3.
Com auxílio de uma ferramenta chamada CEMiTool, desenvolvida por Nakaya com apoio da FAPESP, foi feita uma análise de coexpressão para entender como os genes interagem entre si dentro da rede complexa que existe em cada célula, formando vias de sinalização e vias metabólicas.
“Isso nos permitiu identificar oito principais módulos [genes com perfil similar de resposta] e identificar nessas redes quais são os hubs, ou seja, aqueles genes com maior número de conexões e que, por esse motivo, são os mais promissores alvos a serem explorados na busca por fármacos”, explicou.
O pesquisador ressaltou que todos os dados da pesquisa, tanto os brutos como os obtidos por meio das análises, estão disponíveis em um repositório público e podem ser consultados por qualquer interessado. Também foram disponibilizados os códigos de programação usados no artigo, para que outros possam reproduzir os resultados.
“Nosso trabalho permitiu gerar uma lista de potenciais alvos terapêuticos e, agora, estamos cruzando esses achados com um banco de dados de compostos ativos. Esse cruzamento é feito por meio computacional, mas com base em dados experimentais. Nos orientamos por estudos já publicados, que revelaram drogas capazes de interferir nesses genes de interesse”, disse.
O grupo também continua a análise dos transcritos encontrados nas amostras dos 39 pacientes infectados com chikungunya, agora com foco nos RNAs não codificadores.
Nakaya contou com apoio da FAPESP por meio de um Auxílio à Pesquisa - Regular e um Apoio a Jovens Pesquisadores. A pesquisa também foi apoiada por meio dos CEPIDs Centro de Pesquisa em Toxinas, Resposta Imune e Sinalização Celular (CeTICS) e CRID.
O artigo Systems analysis of subjects acutely infected with the Chikungunya virus, de Alessandra Soares-Schanoski et al, pode ser lido em: journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1007880.
A Agência FAPESP licencia notícias via Creative Commons (CC-BY-NC-ND) para que possam ser republicadas gratuitamente e de forma simples por outros veículos digitais ou impressos. A Agência FAPESP deve ser creditada como a fonte do conteúdo que está sendo republicado e o nome do repórter (quando houver) deve ser atribuído. O uso do botão HMTL abaixo permite o atendimento a essas normas, detalhadas na Política de Republicação Digital FAPESP.
