



Resultados apresentados por grupo do Centro de Inovação em Novas Energias podem tornar o uso de métodos de aprendizado de máquina na ciência de materiais mais assertivo e menos custoso (imagem: CINE/divulgação)
Resultados apresentados por grupo do Centro de Inovação em Novas Energias podem tornar o uso de métodos de aprendizado de máquina na ciência de materiais mais assertivo e menos custoso
Resultados apresentados por grupo do Centro de Inovação em Novas Energias podem tornar o uso de métodos de aprendizado de máquina na ciência de materiais mais assertivo e menos custoso
Resultados apresentados por grupo do Centro de Inovação em Novas Energias podem tornar o uso de métodos de aprendizado de máquina na ciência de materiais mais assertivo e menos custoso (imagem: CINE/divulgação)
Agência FAPESP* – Os programas computacionais de aprendizado de máquina (machine learning) se destacam de todos os outros por terem a capacidade de aprender a partir da experiência, ou seja, a partir da interação com um conjunto de dados. Quanto maior a experiência, melhor é o desempenho desses programas ou modelos na tarefa para a qual foram criados. Contudo, erros acontecem e poder detectá-los e resolvê-los é essencial.
Uma equipe de pesquisadores do Centro de Inovação em Novas Energias (CINE) – um Centro de Pesquisa em Engenharia (CPE) constituído por FAPESP e Shell – realizou um estudo sistemático dos erros cometidos por um modelo de aprendizado de máquina que tinha sido criado para predizer propriedades físico-químicas de um grupo de materiais. O trabalho, publicado no Journal of Chemical Information and Modeling, aumentou o entendimento dos erros e propôs soluções para tornar modelos desse tipo mais precisos.
“Os resultados apresentados podem tornar o uso de métodos de aprendizado de máquina na ciência de materiais mais assertivo e menos custoso”, diz Luis Cesar de Azevedo, um dos autores do artigo que reporta o estudo, que teve apoio da FAPESP.
De fato, existe um interesse crescente no uso de ferramentas de aprendizado de máquina com o objetivo de encontrar materiais ou moléculas que tenham as propriedades desejadas e, portanto, possam cumprir com eficiência determinadas funções em dispositivos ou sistemas. No programa de Ciência Computacional de Materiais e Química (CMSC) do CINE, trabalhos sobre aprendizado de máquina vêm sendo realizados com o objetivo de enfrentar a necessidade de desenvolver ou encontrar materiais eficientes para a geração e o armazenamento de energia.
Para explorar o conjunto praticamente infinito de moléculas possíveis, métodos experimentais, nos quais é necessário sintetizar e caracterizar cada molécula, são impensáveis. Por outro lado, métodos computacionais tradicionais, apesar de serem menos custosos e demorados, tampouco são viáveis em alguns casos. Para ter uma ideia, enquanto simular uma única molécula por um método convencional como a Teoria do Funcional da Densidade pode levar alguns dias, analisar dezenas de milhares de compostos usando um programa de aprendizado de máquina pode tomar poucos segundos.
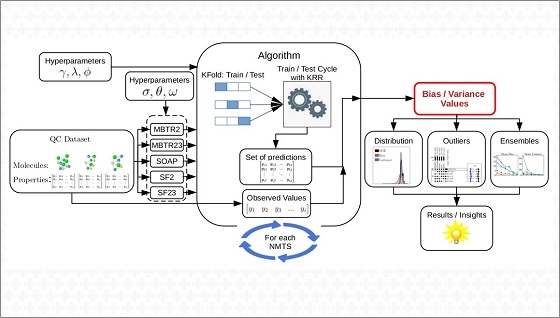
Para isso, é necessário desenvolver um algoritmo (um conjunto de instruções computacionais) e utilizar uma base de dados previamente obtida pela comunidade científica por meio de métodos experimentais ou teóricos. O algoritmo deve, então, fazer seu treinamento, interagindo com os dados e reconhecendo padrões. O resultado dessa experiência é um modelo que será capaz de predizer as propriedades de materiais e moléculas que não constavam na base de dados inicial.
“Apesar de existirem modelos com uma alta acurácia média em alguns domínios, esses modelos podem cometer erros discrepantes [outliers] para algumas moléculas”, explica Azevedo, que é membro do CMSC no CINE. “Este trabalho demonstrou que uma visão detalhada do erro, decompondo-o em erros sistemáticos [viés] e aleatórios [variância], pode mostrar características específicas do desempenho de predição.”
O trabalho também identificou que a maioria dessas imprecisões acontece com moléculas planares (aquelas que possuem ângulos mais abertos e maior distância entre seus átomos).
Felizmente, o artigo mostrou que é possível reduzir os erros utilizando uma combinação de modelos de aprendizado de máquina (ensemble) para predizer as propriedades dos materiais. Além disso, segundo os autores, ao preparar o treinamento do algoritmo é necessário realizar uma seleção mais criteriosa dos dados e dos descritores (os valores computacionais usados para descrever as moléculas do banco de dados).
O estudo foi realizado no âmbito da pesquisa de mestrado em ciência da computação que Azevedo está realizando na Universidade Federal do ABC (UFABC), com a orientação do professor Ronaldo Prati. O trabalho contou com a colaboração de outros membros do CINE: os professores Juarez L. F. Da Silva (IQSC-USP) e Marcos Quiles (Unifesp), e o doutorando Gabriel A. Pinheiro (Unifesp).
O artigo Systematic Investigation of Error Distribution in Machine Learning Algorithms Applied to the Quantum-Chemistry QM9 Data Set Using the Bias and Variance Decomposition pode ser lido em: https://pubs.acs.org/doi/10.1021/acs.jcim.1c00503.
* Com informações da Assessoria de Comunicação do CINE.
A Agência FAPESP licencia notícias via Creative Commons (CC-BY-NC-ND) para que possam ser republicadas gratuitamente e de forma simples por outros veículos digitais ou impressos. A Agência FAPESP deve ser creditada como a fonte do conteúdo que está sendo republicado e o nome do repórter (quando houver) deve ser atribuído. O uso do botão HMTL abaixo permite o atendimento a essas normas, detalhadas na Política de Republicação Digital FAPESP.
