



La neoplasia poco común se detectó en la médula espinal de un paciente de 33 años atendido en el Hospital del Cáncer de la localidad de Barretos, en São Paulo, Brasil (imagen: PLoS One)
Un grupo de investigadores identificó las alteraciones cromosómicas y las mutaciones génicas asociadas al tumor glioneural formador de rosetas del cuarto ventrículo
Un grupo de investigadores identificó las alteraciones cromosómicas y las mutaciones génicas asociadas al tumor glioneural formador de rosetas del cuarto ventrículo
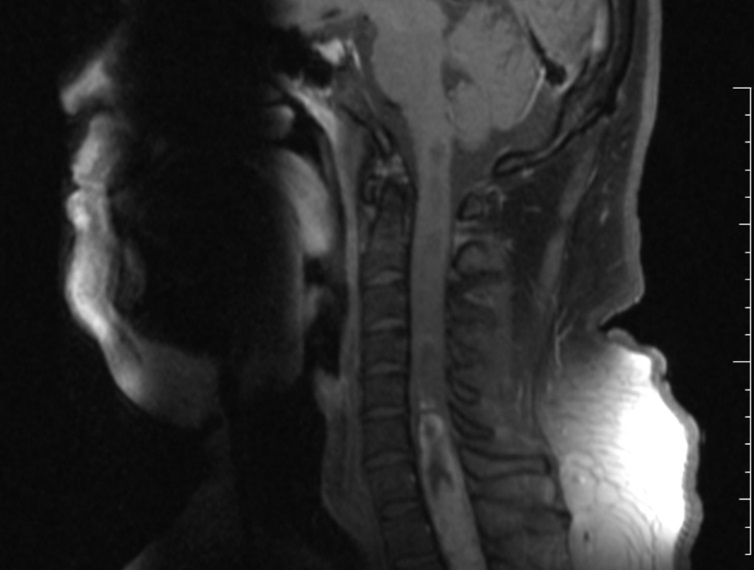
La neoplasia poco común se detectó en la médula espinal de un paciente de 33 años atendido en el Hospital del Cáncer de la localidad de Barretos, en São Paulo, Brasil (imagen: PLoS One)
Por Karina Toledo
Agência FAPESP – Investigadores del Hospital del Cáncer de Barretos (en São Paulo, Brasil) describieron el perfil genético detallado de un tipo raro de cáncer cerebral conocido como tumor glioneural formador de rosetas del cuarto ventrículo (RGNT, por sus siglas en inglés) en la revista PLoS One.
Según Rui Manuel Reis, coordinador de este estudio apoyado por la FAPESP, constan en la literatura científica de 100 casos informados de este tipo de cáncer. Y éste es el primer relato científico del mismo en Brasil.
“Con base en los informes publicados, se sabe que es un tumor indolente, con buen pronóstico y que surge en jóvenes adultos. Hasta ahora se conocían tan sólo algunos marcadores inmunohistoquímicos, y nada más. El objetivo de nuestro estudio consistió en caracterizar este tumor genéticamente, a los efectos de intentar identificar su origen, las alteraciones cromosómicas y las mutaciones involucradas”, dijo Reis.
Este estudio se realizó con el apoyo de la FAPESP durante el posdoctorado de Lucas Tadeu Bidinotto, bajo la supervisión de Reis.
De acuerdo con el investigador, recién en 2007 pasó a considerarse al tumor glioneural formador de rosetas del cuarto ventrículo como una entidad patológica, según el criterio de la Organización Mundial de la Salud (OMS). Antes, en cada relato de caso se utilizaba una nomenclatura distinta. El nombre que ahora es considerado oficial hace referencia al hecho de que constituye un tumor mixto –que abarca tanto neuronas como células de la glía– y también a las estructuras que forman las células tumorales, que se asemejan a los pétalos de una flor.
Mientras que en la mayoría de los casos informados el tumor se ubicó en el cuarto ventrículo cerebral –la cavidad situada cerca de la zona del cerebelo–, el tumor del paciente de 33 años atendido en Barretos se alojaba en la médula espinal. Solamente otros dos eventos de este tipo se habían descrito anteriormente.
Se analizó una muestra del tumor mediante el empleo de distintas metodologías. Con una técnica conocida como microarray de hibridación genómica comparada (CGH, por sus siglas en inglés), el grupo estudió todo el conjunto de cromosomas de las células tumorales en busca de alteraciones.
“Existen cromosomas bastante alterados en ese tumor, en áreas que no son típicas de tumores cerebrales. Hay incrementos en los cromosomas 9 y en el 16, por ejemplo; es decir: en vez de dos copias, encontramos cuatro. También notamos pérdidas en la cantidad de copias del cromosoma 1”, comentó Reis.
Al comparar con el ADN presente en muestras de sangre del mismo paciente, los investigadores descubrieron que las alteraciones se encuentran presentes únicamente en las células tumorales, señal indicativa de que surgieron luego del nacimiento.
“Aunque la mayor parte de las alteraciones encontradas son raras en tumores cerebrales, una que se observó en el cromosoma 7 nos es muy familiar. Se trata de una fusión de dos genes: el BRAF y el KIAA1549”, dijo el investigador. Este hallazgo se confirmó posteriormente mediante la aplicación de otras tres metodologías: la reacción de transcripción reversa seguida de la reacción de cadena de la polimerasa (RT-PCR), la FISH y la secuenciación de ADN.
En un estudio anterior publicado en Journal of Neuropathology and Experimental Neurology, el grupo demostró que esa fusión se encuentra en el 60% de los casos de astrocitoma pilocítico (un tipo de tumor cerebral originado en células cerebrales denominadas astrocitos, uno de los más frecuentes en niños) y que su presencia está asociada a un buen pronóstico (lea más en http://agencia.fapesp.br/21930).
“Este hallazgo nos permite formular la hipótesis de que el tumor glioneural formador de rosetas tendría alguna relación etiológica con el astrocitoma pilocítico, que también es un tumor indolente que afecta a jóvenes”, dijo Reis.
Genes mutados
Para evaluar el perfil de mutaciones existentes se utilizó la secuenciación de nueva generación, inicialmente con un panel de 20 genes blanco creado en el marco de un proyecto FAPESP orientado al estudio de tumores gliales. Luego se procedió a la secuenciación del exoma, que abarca todos los genes codificadores de proteínas (el 2% del genoma humano).
“Ninguno de los 20 genes blanco había mutado, entonces decidimos hacer la secuenciación en gran escala de todos los genes codificadores de proteínas. Encontramos cuatro mutaciones somáticas (adquiridas después del nacimiento y presentes únicamente en el tejido tumoral), que después se confirmaron mediante la aplicación del método de secuenciación tradicional”, comentó Reis.
Entre los genes mutados estaban el SCN1A, asociado a formas hereditarias de epilepsia, y el MLL2, al que recientemente se lo relacionó con el desarrollo de meduloblastoma, y que parece regular la forma de expresión de los genes mediante mecanismos epigenéticos tales como la metilación del ADN.
También se hallaron otros dos genes mutados acerca de los cuales no había nada descrito en la literatura científica: el CNNM3 y el PCDHGC4.
“Ahora sería interesante validar los resultados en otros tumores del mismo tipo para intentar comprobar si efectivamente los cuatro genes están implicados en el surgimiento del RGNT. Pretendemos entrar en contacto con investigadores que informaron sobre la detección de este tipo de tumor y ver si existen posibilidades de estudios en colaboración”, dijo Reis.
Puede leerse el artículo Molecular Profiling of a Rare Rosette-Forming Glioneuronal Tumor Arising in the Spinal Cord (doi: 10.1371/journal.pone.0137690) en la siguiente dirección: journals.plos.org/plosone/article?id=10.1371/journal.pone.0137690.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
