



Pesquisadores do Centro de Estudos do Genoma Humano e Células-Tronco da USP reconstituem processo celular da síndrome de Richieri-Costa Pereira (esq: células de crista neural geradas a partir de iPSCs de pacientes com sindrome de Richieri-Costa-Pereira/imagem: Gerson Kobayashi; dir: células de crista neural - imunofluorescência para vimentina e p75, expressos neste tipo de célula/imagem: Luiz Caires
Pesquisadores do Centro de Estudos do Genoma Humano e Células-Tronco da USP reconstituem processo celular da síndrome de Richieri-Costa Pereira
Pesquisadores do Centro de Estudos do Genoma Humano e Células-Tronco da USP reconstituem processo celular da síndrome de Richieri-Costa Pereira
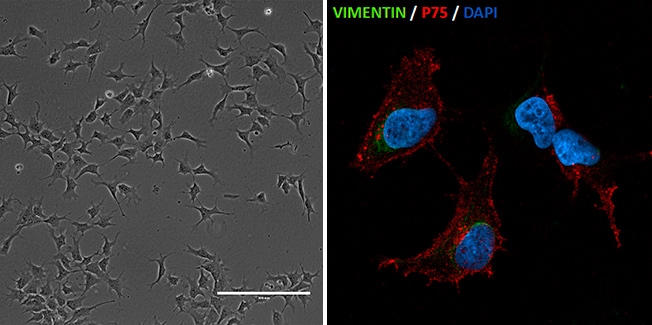
Pesquisadores do Centro de Estudos do Genoma Humano e Células-Tronco da USP reconstituem processo celular da síndrome de Richieri-Costa Pereira (esq: células de crista neural geradas a partir de iPSCs de pacientes com sindrome de Richieri-Costa-Pereira/imagem: Gerson Kobayashi; dir: células de crista neural - imunofluorescência para vimentina e p75, expressos neste tipo de célula/imagem: Luiz Caires
Maria Fernanda Ziegler | Agência FAPESP – Os mecanismos celulares e de expressão gênica que levam à má-formação da mandíbula, traço mais característico da síndrome de Richieri-Costa Pereira, acabam de ser esclarecidos.
Pesquisadores do Centro de Pesquisa sobre o Genoma Humano e Células-Tronco (CEGH-CEL) e da Duke University, nos Estados Unidos, descobriram que nos casos da síndrome ocorrem problemas no processo de migração e diferenciação celular durante a formação do crânio e face, processos que ocorrem no primeiro trimestre de gestação humana.
A síndrome de Richieri-Costa Pereira é uma doença autossômica recessiva descrita pela primeira vez em 1992 pelo professor Antonio Richieri-Costa e equipe do Hospital de Reabilitação de Anomalias Craniofaciais da Universidade de São Paulo (USP) em Bauru. Atualmente há mais de 20 pacientes conhecidos com a doença no Brasil (e um no exterior), mas o estudo da doença pode dar pistas importantes sobre a formação craniofacial durante o desenvolvimento fetal e até mesmo para a bioengenharia.
Os pesquisadores do CEGH-CEL – um Centro de Pesquisa, Inovação e Difusão (CEPID) da FAPESP sediado na USP – já haviam descoberto, em 2014, que mutações no gene EIF4A3 estão relacionadas a anomalias decorrentes da síndrome. Um artigo recém-publicado na Human Molecular Genetics descreve os mecanismos celulares que desencadeiam a má-formação ao reconstituir as etapas do desenvolvimento craniofacial.
A formação da cabeça é um processo complexo, com várias etapas e processos envolvidos. Para que ela ocorra é preciso haver proliferação, migração celular e também a morte celular programada, chamada de apoptose. Os pesquisadores observaram que pacientes com a síndrome apresentam problema de migração celular durante o processo de formação craniofacial.
“Se houver problema de migração, haverá menos células povoando os tecidos que serão formados – por exemplo, a mandíbula e outros componentes do complexo craniofacial. Achamos que a quantidade reduzida de células devido à migração defeituosa resultaria em menos células para gerar esses tecidos”, disse Gerson Kobayashi, um dos autores do estudo cuja tese de doutorado foi a respeito do tema.
A reconstituição in vitro das etapas do desenvolvimento craniofacial foi feita a partir de células somáticas – células-tronco da polpa do dente ou fibroblasto ou ainda células do sangue – retiradas de pacientes com a síndrome de Richieri-Costa Pereira.
Essas células foram então reprogramadas para virar células pluripotentes, parecidas com células-tronco embrionárias. Isso permitiu criar células de crista neural (responsáveis por formar a maior parte dos ossos e cartilagens do complexo craniofacial) e células que depois de atingir um alto grau de diferenciação viram mesenquimais (células com plasticidade elevada que podem originar vários tecidos).
“Com as células de crista neural, conseguimos observar esses três processos e identificar se algum deles está comprometido nos pacientes. Com a proliferação e apoptose estava tudo bem, mas na migração não. O importante é que a célula de crista neural ao chegar a uma determinada região vai se diferenciar. Testamos a diferenciação para cartilagem e tecido ósseo e confirmamos que os dois processos estão comprometidos”, disse a professora Maria Rita Passos-Bueno, coordenadora do Laboratório de Diagnóstico Molecular de Doenças do CEGH-CEL.
Melhor prognóstico de cirurgia
Os estudos in vitro realizados no CEGH-CEL foram replicados em modelo animal por pesquisadores da Duke University. Kobayashi estima que será possível usar o modelo para procurar drogas que revertam o fenótipo da célula e possam ser utilizadas em gravidez de risco. A ideia é que os resultados do estudo possam servir de base para cirurgias mais eficientes.
“Vimos que há capacidade de formar osso, mas ela ocorre na época errada. A próxima pergunta a responder é se será possível intervir na hora de fazer uma cirurgia, porque esses pacientes precisam passar por vários procedimentos cirúrgicos. Com isso, seria possível administrar substâncias para que ocorra um processo de reconstituição óssea mais eficiente, ajudando a melhor prognóstico da cirurgia”, disse Passos-Bueno.
A pesquisadora estima que descobertas sobre os mecanismos da síndrome Richieri-Costa Pereira possam ser relevantes para estudos em bioengenharia de tecidos.
“O gene EIF4A3 parece ter um papel importante na formação da mandíbula e há um investimento para a produção de estruturas ósseas em laboratório, que poderão então ser usadas para substituição de tecidos ósseos comprometidos. Para isso, é preciso conhecer quais são as moléculas críticas, por exemplo, para garantir que a mandíbula tenha um determinado tamanho. A aplicação do conhecimento a longo prazo é para os pacientes, mas também para a bioengenharia de tecidos”, disse Passos-Bueno.
O artigo EIF4A3 deficient human iPSCs and mouse models demonstrate neural crest defects that underlie Richieri-Costa-Pereira syndrome, de Emily E. Miller, Gerson S. Kobayashi, Camila M. Musso, Miranda Allen, Felipe A.A. Ishiy, Luiz Carlos de Caire Jr., Ernesto Goulart, Karina Griesi-Oliveira, Roseli M. Zechi-Ceide, Antonio Richieri-Costa, Debora R. Bertolam, Maria Rita Passos-Bueno e Debra L. Silver, pode ser lido em: https://doi.org/10.1093/hmg/ddx078.
A Agência FAPESP licencia notícias via Creative Commons (CC-BY-NC-ND) para que possam ser republicadas gratuitamente e de forma simples por outros veículos digitais ou impressos. A Agência FAPESP deve ser creditada como a fonte do conteúdo que está sendo republicado e o nome do repórter (quando houver) deve ser atribuído. O uso do botão HMTL abaixo permite o atendimento a essas normas, detalhadas na Política de Republicação Digital FAPESP.
