



The technique was used by researchers affiliated with institutions in Brazil, Germany and Finland to study the SARS-CoV-2 main protease (Mpro), a key driver of the virus’s reproductive cycle (clockwise: illustration of SARS-CoV-2 Mpro dimer structure; computer simulation of inhibitor N1 interacting with Mpro; two-dimensional representation of N1, highlighting interactions with Mpro active sites/Journal of Biomolecular Structure and Dynamics)
The technique was used by researchers affiliated with institutions in Brazil, Germany and Finland to study the SARS-CoV-2 main protease (Mpro), a key driver of the virus’s reproductive cycle.
The technique was used by researchers affiliated with institutions in Brazil, Germany and Finland to study the SARS-CoV-2 main protease (Mpro), a key driver of the virus’s reproductive cycle.
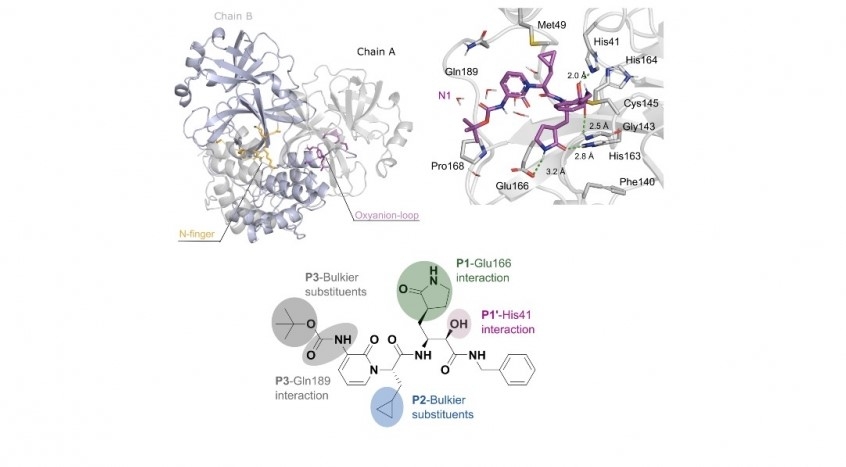
The technique was used by researchers affiliated with institutions in Brazil, Germany and Finland to study the SARS-CoV-2 main protease (Mpro), a key driver of the virus’s reproductive cycle (clockwise: illustration of SARS-CoV-2 Mpro dimer structure; computer simulation of inhibitor N1 interacting with Mpro; two-dimensional representation of N1, highlighting interactions with Mpro active sites/Journal of Biomolecular Structure and Dynamics)
By Janaína Simões | Agência FAPESP – A study by researchers affiliated with institutions in Brazil, Germany and Finland proposes a new standard for computer simulation that promises to accelerate the search for novel bioactive compounds against the virus that causes COVID-19. The researchers used the procedure to analyze a key protein in the reproductive cycle of SARS-CoV-2, which has been a major focus of attention for scientists and the pharmaceutical industry as a target for antiviral drugs. They estimate that the method they have developed can cut the time taken in the initial stage of basic research from two or three years to under a year.
An article reporting the results of the study is published in the Journal of Biomolecular Structure and Dynamics. The authors explain that the outer protein layer of SARS-CoV-2 is rich in the amino acid cysteine, which must be active and intact for the virus to remain viable, and is therefore referred to as its main protease (Mpro). Proteases are enzymes that cleave the peptide bonds between amino acids and proteins and break down polyproteins (protein chains) into smaller proteins, which in this virus are used to produce the RNA that encodes key structures such as the spike (the component that enables it to invade and infect human cells) and the viral envelope (the outer layer that protects its genetic material).
Mpro is considered a target for drugs against COVID-19 because inhibiting cleavage of key proteins could block viral invasion and replication. One of the potential strategies entails synthesizing chemical components designed to bind to specific parts of such proteins so as to inactivate them.
Drug discovery and development takes about 20 years on average. The study just published proposes a method that could reduce this window by a year in the case of novel bioactive compounds against SARS-CoV-2. The search for active compounds is the initial stage of drug discovery and typically takes two to three years.
“The pre-clinical phase of the development of the vaccines the world is now administering was accelerated by the use of published information about SARS-CoV-1, from which SARS-CoV-2 evolved. We can’t benefit in this way while doing basic research to develop a drug because we lack the requisite base information,” said Glaucio Monteiro Ferreira, first author of the article.
Ferreira is a professor in the Clinical and Toxicological Analysis Department at the University of São Paulo’s School of Pharmaceutical Sciences (FCF-USP) in Brazil and conducted part of the research while he was a postdoctoral fellow at Tübingen University Hospital in Germany, with FAPESP’s support (16/12899-6 and 19/24112-9).
Another reason for the long lead time in drug development is the complexity of researching how best to administer the substance (orally or by injection, for example). “If it is a medication to be ingested, we have to make sure it will pass through all the barriers in the body to reach the site where it should act. These details explain why drugs take longer to develop than vaccines,” he said.
Dimers
Ferreira used advanced bioinformatics and structural biology techniques to investigate Mpro (also called 3CLpro), which needs cysteine as a substrate or “food” to perform its function. Previous research had shown that many key proteins in SARS-CoV-2 are monomers, meaning they have a single chain of amino acids. “However, we know the virus is a dimer, with doubled protein chains. This complicates drug discovery because you have to find a compound that can prevent formation of both chains,” he said.
Promising results had been achieved using covalent cysteine-binding inhibitors in previous research. In this study, the aim was to find a means of inhibiting cysteine itself so as to prevent Mpro from “feeding” on it and block viral replication.
Computer simulations were run to look for a compound that prevented formation of the two chains, using molecular dynamics to analyze the physical motion of atoms in a simulated viral attack on human cells. Ferreira and his group discovered from the simulations with one and two protein ligands that analysis of monomers using X-ray diffraction captured results after cysteine consumption and cleavage of the protein, an approach that fails to focus on blocking of this phase in protein replication, the process of interest.
They then simulated the use of two inhibitors that affect the action of Mpro – covalently bound ligands N1 and N3 – and found the former to be more effective in that it did not permit electric charge donation to cysteine, “starving” the enzyme as a result.
“Our simulations led us more quickly to this inhibitor, which really can block the action of the enzyme, indicating the compound’s potential to become a powerful drug,” Ferreira said. Coincidentally, Pfizer recently announced an initiative to find a COVID-19 drug targeting Mpro, although Ferreira’s research began long ago.
His collaborators were Thales Kronenberger and Antti Poso at Tübingen University Hospital (Germany); Arun Kumar Tonduru at the University of Eastern Finland; and Rosario Dominguez Crespo Hirata and Mario Hiroyuki Hirata at the University of São Paulo (USP) in Brazil. Investigation was performed during Ferreira’s postdoctoral research at the Molecular Biology Laboratory for Diagnosis and Farmacogenomics (LBMAD) under supervision of Professor Hirata.
The article “SARS-COV-2 Mpro conformational changes induced by covalently bound ligands” is at: www.tandfonline.com/doi/full/10.1080/07391102.2021.1970626.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
