




Pancreatic islet from a mouse treated with STZ, antibiotic and MDP showing markedly low numbers of cells stained for insulin, indicating destruction of insuling-producing beta cells (image: FMRP-USP)
Brazilian study suggests that bacterial components may activate innate immune system receptors and contribute to the development of adaptive response to insulin-producing beta cells
Brazilian study suggests that bacterial components may activate innate immune system receptors and contribute to the development of adaptive response to insulin-producing beta cells
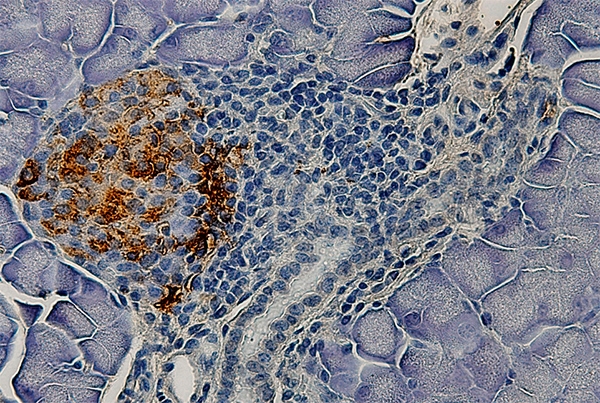
Pancreatic islet from a mouse treated with STZ, antibiotic and MDP showing markedly low numbers of cells stained for insulin, indicating destruction of insuling-producing beta cells (image: FMRP-USP)
By Karina Toledo | Agência FAPESP – Recent research has shown that diabetics often exhibit an imbalance between beneficial and pathogenic bacteria in the intestinal microbiota. This disruption of the gut microbial community is potentially harmful to the organism, but it is not clear whether it is one of the causes or consequences of diabetes.
New evidence published by Brazilian researchers in the Journal of Experimental Medicine suggests that intestinal bacteria can escape into the lymphatic ganglia located near the pancreas owing to alterations in the intestinal wall’s permeability caused by dysbiosis. These bacteria may then activate certain receptors in cells of the innate immune system (the organism’s first line of defense), and especially macrophages and dendritic cells.
According to the authors, this activation induces a pro-inflammatory condition in the organism and favors an adaptive immune response targeting the insulin-producing beta cells in the pancreas. This process results in type 1, or autoimmune, diabetes.
“The last stage of type 1 diabetes is well understood. We know that at some point, the immune system starts attacking pancreatic beta cells as if they were foreign to the organism,” said Daniela Carlos, a researcher at the University of São Paulo’s Ribeirão Preto Medical School (FMRP-USP) and principal investigator for the FAPESP-funded research project. “As a result, specific cells known as T lymphocytes are activated, and antibodies are produced to destroy insulin-producing cells. However, it isn’t clear what triggers the innate immune system to induce this adaptive immune response. Our study shows an intracellular receptor called NOD2 is involved.”
The findings presented in the article are based on experiments with mice performed during Frederico R. C. Costa’s master’s research, supervised by Carlos and with the collaboration of João Santana Silva, a professor at FMRP-USP.
In the experiments, the researchers observed that mice that had been genetically modified so as not to express the protein NOD2 were resistant to the development of type 1 diabetes, even when challenged with a chemical stimulus.
“Diabetes was induced in healthy rodents in the laboratory by administering a drug called streptozotocin, which is toxic to pancreatic beta cells,” Carlos said. “The death of these cells acts as an inflammatory signal. Other defense cells are activated and recruited to the site, where they recognize and attack insulin-producing beta cells. The animals received the substance for five consecutive days. Fifteen days later, they were already diabetic.”
The disease was confirmed by tests for parameters such as fasting blood sugar, glucose tolerance, and insulin tolerance. All these clinical parameters remained unaltered in the animals that received streptozotocin but did not express NOD2.
Suspect’s profile
This receptor’s role is well described in the scientific literature, Carlos explained. NOD2 is present in defense cells and the intestinal epithelium, where its role is to recognize muramyl dipeptide (MDP), a component of bacterial cell walls. When activated, the receptor induces intracellular signaling that results in the expression of antimicrobial peptides and inflammatory cytokines such as interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and interleukin-23 (IL-23), substances involved in the activation of defense cells and their migration to the intestine. NOD2 plays a key role in local and systemic immunity by maintaining the integrity of the intestinal barrier and controlling bacterial translocation from the lumen to the mucosa.
“We decided to investigate how NOD2 participates in the pathogenesis of type 1 diabetes because although previous research had found a link between altered microbiota and autoimmune diabetes in humans and in experimental models, the mechanisms by which intestinal bacteria leads to development of the disease remained obscure,” Carlos said. “In this context, NOD2 seemed to be a key target for study in light of its role in maintaining intestinal homeostasis.”
Autoantigens are constantly being presented to T lymphocytes by cells of the innate immune system, she went on. This process is normally tolerogenic: through interleukin-10 (IL-10), the innate immune system signals that T lymphocytes should play a regulatory (immunosuppressive) role.
According to the Ribeirão Preto group’s theory, however, when bacterial components activate the receptor NOD2 in dendritic cells and macrophages, releasing inflammatory cytokines, the T lymphocytes may receive a different signal because of the inflammation and become pathogenic: that is, capable of recognizing and attacking insulin-producing beta cells.
In summary, pancreatic beta cell death followed by the release of autoantigens is necessary to create the inflammatory context. In laboratory animals, cell death is induced by streptozotocin. In humans, the cause can be an environmental factor such as viral infection, Carlos explained.
Validation
A second experiment was performed with mice to validate NOD2’s importance in the pathogenesis of type 1 diabetes and confirm the participation of intestinal bacteria in its activation.
This time, the intestinal microbiota was reduced in a group of mice using a potent cocktail of antibiotics. When the mice in this group were challenged with streptozotocin, they did not develop disease, even though NOD2 was expressed. This effect was due to the elimination of bacteria in the pancreatic lymph nodes, according to the researchers.
In another group of mice, the intestinal microbiota was also reduced, but in addition to streptozotocin, the mice were given injections of MDP, a NOD2-activating molecule found in several bacteria. These animals became diabetic.
“These findings confirm that bacteria recognized via NOD2 in the pancreatic lymph nodes are involved in the development of type 1 diabetes,” Carlos said. “We haven’t discovered whether one or more bacteria are involved and haven’t identified the species, but we now plan to perform a more comprehensive metagenomic analysis with the aim of doing so.”
Based on this evidence, she added, the next step will be to test a number of preventive or therapeutic interventions, such as modulation of the intestinal microbiota using probiotic and prebiotic compounds or drug-induced inhibition of NOD2.
The results published in the Journal of Experimental Medicine were also presented by Daniela Carlos during the FAPESP/EU-LIFE Symposium on Cancer Genomics, Inflammation & Immunity, held on June 7-9 at FAPESP’s headquarters in São Paulo to bolster collaboration between scientists in São Paulo State and Europe.
The article “Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset” (doi: 10.1084/jem.20150744) can be read at http://jem.rupress.org/content/213/7/1223.abstract.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
