



In an article published in the journal Chemistry & Biology, researchers describe a new mechanism that inhibits the activity of proteasomes, protein complexes that are a target for cancer therapy
In an article published in the journal Chemistry & Biology, researchers describe a new mechanism that inhibits the activity of proteasomes, protein complexes that are a target for cancer therapy.
In an article published in the journal Chemistry & Biology, researchers describe a new mechanism that inhibits the activity of proteasomes, protein complexes that are a target for cancer therapy.
In an article published in the journal Chemistry & Biology, researchers describe a new mechanism that inhibits the activity of proteasomes, protein complexes that are a target for cancer therapy
By Karina Toledo
Agência FAPESP – A new mechanism to inhibit proteasomes, protein complexes that are a target for cancer therapy, is the topic of an article published in the journal Chemistry & Biology. The first author of the study is Brazilian Daniela Trivella, researcher at the Brazilian Biosciences National Laboratory at the Brazilian Center for Research in Energy and Materials (LNBio/CNPEM).
The findings of the study, conducted with FAPESP support in partnership with researchers from the University of California in San Diego, United States, and at the Technische Universität München, in Germany, are paving the way for the development of a new generation of chemotherapy drugs that are more effective and less toxic.
“We have already developed a series of molecules based on the newly identified mechanism. Now we plan to synthesize them in partnership with CNPEM researcher Marjorie Bruder and test their potential. The goal is to optimize the proteasome inhibition effect, make the compound even more selective of tumor cells and eliminate the resistance problems found with drugs that are currently available on the market,” Trivella said.
A member of the category of enzymes known as proteases, the proteasome is a protein complex responsible for several essential functions inside cells, such as eliminating harmful or non-functioning proteins and regulating the processes of apoptosis (programmed cell death), cell division and proliferation.
Since approval of the drug bortezomib in 2003, it has been demonstrated that molecules capable of inhibiting the activity of the proteasome kill tumor cells more effectively and have a less severe impact on healthy cells.
“Cancer cells are more dependent on proteasome than other cells because cancer cells have an accelerated metabolism and a high rate of division and proliferation and thus live in a chronic state of oxidative and protein stress. They very quickly end up synthesizing excess low-quality proteins. The proteasome is the big garbage collector that tries to clean up the mess in the tumor cell,” Trivella explained.
The researcher went on to say that by inhibiting the proteasome, we know that it is possible to increase the rates of apoptosis and reduce the rate of cell division and proliferation, which are altered in the tumor.
Although bortezomib has been used successfully since it was approved, mainly in treating multiple myeloma (bone marrow cancer), the drug presents significant adverse side effects, including peripheral neuropathy, which is characterized by tingling, pain or numbness in the arms and legs.
According to Trivella, this occurs because chemotherapy inhibits not only the functioning of the proteasome but also that of other proteases that are important to the body.
In 2012, the drug carfilzomib, inspired by a natural molecule called epoxomicin, was approved. Carfilzomib is more selective and effective against cancer; however, because it irreversibly inhibits proteasomes both in tumor cells and healthy cells, the drug may cause prolonged use to be harmful due to high toxicity.
Over time, Trivella explained, certain mutations in proteasomes and alternative biochemical mechanisms were observed, causing tumors to become resistant to the drugs available on the market. In addition, the first generation of inhibitors did not prove to be very effective in treating solid tumors, which limits their application.
In search of alternatives
Also in 2012, U.S. and Brazilian researchers isolated a natural molecule in cyanobacteria from the Caribbean called carmaphycin, whose reactive group (the portion of the molecule that interacts with the proteasome) is the same as that of carfilzomib. The molecule is known as an epoxyketone.
“Epoxyketones are very potent selective inhibitors of the proteasome because they interact with this enzyme in two stages: the first reversible and the second irreversible,” Trivella explained.
However, in addition to irreversibly inhibiting proteasomes, the compound presented certain chemical instability problems. To optimize its effect and find new reactive groups, researchers from the Scripps Institution of Oceanography at the University of California in San Diego developed a series of synthetic analogs with slight structural modifications.
Trivella tested these compounds during an internship in California in her post-doctoral research when she was still associated with the Chemistry Institute at the University of Campinas (Unicamp).
“My task was to compare the potency of the various compounds, and if we found something interesting, we would study it further. To do this, response concentration curves were made, which determined the drug concentration necessary to cause 50% deactivation of the proteasome (IC50) or, in parallel experiments, the death of half of the tumor cells in culture,” the researcher explained.
One of the molecules tested had an enone as a reactive group and had characteristics of carmaphycin and another natural molecule named syringolin, isolated from plant pathogens.
“Syringolin had already been reported as an irreversible inhibitor of the proteasome in a single-step reaction with an IC50 value in the micromolar order, which is not very potent. We wanted to determine the effects of combining the chemical skeleton of carmaphycin with reactive groups more similar to those of syringolin. We had thought that this new molecule with an enone reactive group would be very similar, but it was 10 times more potent than syringolin,” Trivella said.
By investigating the reaction mechanisms of the new molecule, named carmaphycin-syringolin enone, the researcher verified that unlike syringolin, and thus like the epoxyketone, the enone interacts with the proteasome in two stages, with the second stage being irreversible.
Additionally, Trivella had observed that in the case of the enone, the second reaction occurs more slowly, increasing the duration of the reversible phase of carmaphycin-syringolin enone inhibition.
“Because the irreversible inactivation of the proteasome has toxic effects, the best window of reversibility observed for the carmaphycin-syringolin enone will potentially reduce the toxicity of this new class of proteasome inhibitors,” Trivella said. “The compound would therefore present a balance between selectivity and potency.”
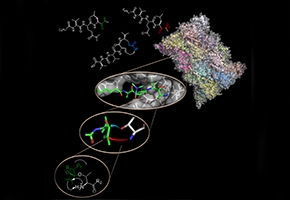
Toxicity tests are still underway, says Trivella. In parallel, studies have been conducted with the help of crystallography techniques (consisting of making crystals from concentrated solutions of the target enzyme in purified form and then studying them through X-ray diffraction) to discover exactly how the interaction between the enzyme target and the carmaphycin- syringolin enone target occurs.
“We discovered that a chemical reaction called hydroamination occurs, which had never before seen under physiological conditions. This type of reaction is frequently used by synthetic chemists in preparing substances, but normally it requires very specific temperature and pH conditions and the use of catalysts to occur. It has never been reported as a mechanism of enzyme inhibition,” Trivella said.
Inspired by this new mechanism for proteasome inhibition, the LNBio group plans to synthesize and test a new series of carmaphycin-syringolin enone analogs to determine their effects on the therapeutic window (preferential death of tumor cells in relation to healthy cells) and assess whether they are also capable of reacting with proteasomes that are resistant to traditional inhibitors.
Another of Trivella’s goals is to look for natural compounds in Brazilian biodiversity that could serve as inspiration for the design of other categories of proteasome inhibitors.
“We need a little inspiration from nature to expand the options of reactive groups and molecule structures. By analyzing the chemical diversity available in our biodiversity, we’ll be able to increase our chances of finding even more innovative alternatives for optimizing properties such as potency, selectivity and pharmacokinetics,” she said.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
