




With aging, mutant genetic material tends to build up in the organelles responsible for producing energy, and this can lead to disease. Brazilian researchers have discovered that a cell cleansing mechanism known as autophagy can modulate this phenomenon (mitochondria being engulfed by autophagosome; credit: Júlio C. B. Ferreira/USP)
With aging, mutant genetic material tends to build up in the organelles responsible for producing energy, and this can lead to disease. Brazilian researchers have discovered that a cell cleansing mechanism known as autophagy can modulate this phenomenon.
With aging, mutant genetic material tends to build up in the organelles responsible for producing energy, and this can lead to disease. Brazilian researchers have discovered that a cell cleansing mechanism known as autophagy can modulate this phenomenon.
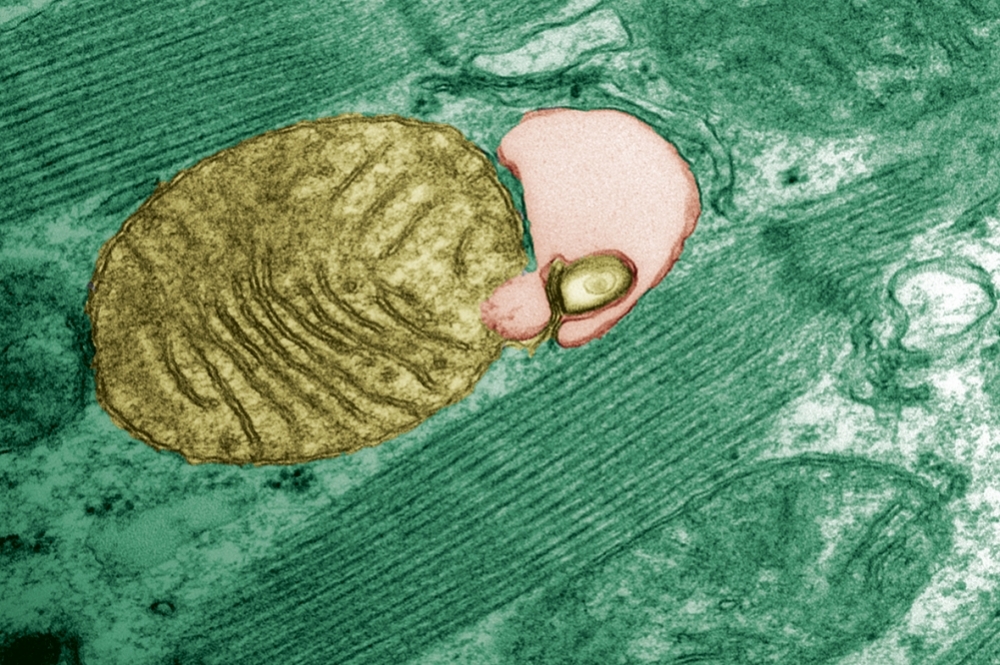
With aging, mutant genetic material tends to build up in the organelles responsible for producing energy, and this can lead to disease. Brazilian researchers have discovered that a cell cleansing mechanism known as autophagy can modulate this phenomenon (mitochondria being engulfed by autophagosome; credit: Júlio C. B. Ferreira/USP)
By Theo Ruprecht | Agência FAPESP – A study by Brazilian scientists points to the probable biological mechanism that modulates the accumulation of mutant mitochondrial DNA in cells during aging, especially in the liver. The mechanism is called autophagy, a process of cellular cleansing that destroys organelles, including mitochondria, and recycles their components. The findings resolve a paradigm in the field and raise a hypothesis for the emergence of diseases associated with mitochondrial DNA, paving the way for the development of therapies.
The study was supported by FAPESP and is reported in an article published in the journal Autophagy.
Mitochondria are responsible for cellular respiration, processing substrates to supply energy to the organism. They have their own genome inherited solely from the mother, unlike the DNA found in cell nuclei, which is inherited from both parents. Mitochondrial DNA is also subject to mutations, some of which can lead to diseases. According to estimates, disorders caused by mitochondrial DNA mutations affect at least one in every 5,000 people worldwide. One of these disorders is MELAS syndrome (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes), which can cause seizures, pain, altered consciousness and focal neurological deficits, among other symptoms.
“Besides the disorders due directly to mitochondrial DNA mutations, there are others to which mutations contribute at least partly,” said Marcos Chiaratti, last author of the article. These include diabetes, Parkinson’s and Alzheimer’s, he added, indicating that the study could pave the way for treatments involving autophagy in the context of different diseases.
Chiaratti is a professor in the Department of Genetics and Evolution at the Federal University of São Carlos (UFSCar) in São Paulo state. The other authors of the article are researchers affiliated with UFSCar and the University of São Paulo (USP) in Brazil, and with Cedars-Sinai Medical Center and Stanford University in the United States. The study was led by Chiaratti and supported by FAPESP via seven projects (16/07868-4, 17/05899-2, 17/04372-0, 18/20028-0, 20/15412-6, 19/25049-9, and 12/50231-6).
Aging
Research has shown that mitochondrial DNA mutations build up as part of aging. “One of the oldest questions in biomedicine is why this is so,” Chiaratti said. Theories that blame mitochondrial DNA replication or cell proliferation have been ruled out by previous studies. Given the importance of autophagy in the life cycle of cells and mitochondrial recycling, Chiaratti and colleagues decided to see if it played a role in mitochondrial DNA mutations. “Autophagy is in vogue. The scientific literature on the topic is growing,” he said. The 2016 Nobel Prize in Physiology or Medicine was awarded to Japanese cell biologist Yoshinori Ohsumi for his discoveries relating to autophagy mechanisms.
Methodology
Practically all humans have a small amount of mutant DNA. The coexistence of mutant and wild-type (non-mutant) mitochondrial DNA in a cell is called heteroplasmy. In this study, Chiaratti et al. used mice with heteroplasmy at 30%: a mutant strain known as NZB/BINJ (or NZB) accounted for 30% of their mitochondrial genome, while the rest was C57BL/6N (or BL6), considered “normal”.
The researchers cross-bred these mice with others that lacked the gene atg7 specifically in the liver. Without this gene, the requisite machinery to make autophagy possible simply disappears. “We spent almost a year just on breeding,” Chiaratti said.
After this period, they had two groups of mice: a control group with NZB mitochondrial DNA and the gene atg7; and a group with NZB but without the gene atg7 in liver cells, so that autophagy in the liver was impossible (liver-specific atg7 knockout).
Experiments were conducted to see if autophagy affected the buildup of mutant mitochondrial DNA over time. Ten days after birth, similar levels of NZB were found in the tissue of both groups of mice, but after 21 days NZB began to accumulate in the livers of the control animals, while the knockout group had the same proportion as before. After 100 days, when mice are considered adults, the controls had even higher levels of NZB while the knockouts had practically the same levels as when they were born, making the difference between the two groups even more evident and suggesting that autophagy plays a role in the age-related buildup of mutant mitochondrial DNA.
The researchers then set out to validate this finding by conducting an experiment with mice that lacked the gene Prkn, which is responsible for a specific stage of autophagy. In Prkn knockout mice, autophagy occurred but was defective. “We again compared these mice with the controls,” Chiaratti said. Although the Prkn knockout mice did eventually accumulate NZB mitochondrial DNA, the buildup was smaller than for the control group. “This reinforced the theory that autophagy is involved since manipulation of another gene associated with it led to changes in the data.”
The researchers also found that autophagy mostly cleared BL6 mitochondrial DNA in controls, which presumably explained the higher levels of NZB. On exploring this possibility further, they discovered that the presence of NZB enhanced mitochondrial respiratory capacity in liver cells. “We, therefore, postulated that autophagy might help eliminate less functional mitochondria while conserving the fittest, which of course would be positive,” Chiaratti said.
Another stage of the study entailed isolation of fibroblasts, a type of connective tissue cell, from the mice, and the in vitro observation that NZB mitochondrial DNA levels in them fell over time. However, when drugs that stimulated autophagy were applied, the decline was reversed. “This was further evidence of the importance of autophagy to mitochondrial DNA regulation,” Chiaratti said.
Next steps
While NZB was considered mutant in the mice involved in these experiments, the results show that it is not detrimental to the liver. On the contrary, it enhances mitochondrial respiratory capacity in liver cells, as already noted. “Now we need to investigate the phenomenon with different mitochondrial DNAs that are known to be pathological,” Chiaratti said.
If autophagy also proves relevant in this context, the researchers can begin to test treatments that stimulate the cell cleansing mechanism against diseases affected by the functioning of mitochondria. Other possibilities for future research, Chiaratti added, will include the role of autophagy in mitochondrial DNA accumulation in tissue elsewhere in the body apart from the liver and isolated fibroblasts.
The article “Autophagy deficiency abolishes liver mitochondrial DNA segregation” is at: www.tandfonline.com/eprint/76MFBNGZFGNQQQYYR2TX/full?target=10.1080/15548627.2022.2038501.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
