

Mediante un abordaje de biología de sistemas, científicos brasileños identificaron diversos genes que podrán explotarse como blancos terapéuticos y como biomarcadores de predisposición a la artralgia crónica (imagen: PLOS Pathogens y kjpargeter/ Freepik)
Mediante un abordaje de biología de sistemas, científicos brasileños identificaron diversos genes que podrán explotarse como blancos terapéuticos y como biomarcadores de predisposición a la artralgia crónica
Mediante un abordaje de biología de sistemas, científicos brasileños identificaron diversos genes que podrán explotarse como blancos terapéuticos y como biomarcadores de predisposición a la artralgia crónica
Mediante un abordaje de biología de sistemas, científicos brasileños identificaron diversos genes que podrán explotarse como blancos terapéuticos y como biomarcadores de predisposición a la artralgia crónica (imagen: PLOS Pathogens y kjpargeter/ Freepik)
Por Karina Toledo | Agência FAPESP – Herramientas computacionales aplicadas a la biología están revolucionando el modo de estudiar lo que ocurre en el interior de las células durante una infección, ayudando así a develar el mecanismo de acción de enfermedades y a hallar potenciales dianas terapéuticas.
Tal es el caso de un trabajo publicado recientemente en la revista PLOS Pathogens, en el cual científicos brasileños analizaron células sanguíneas de pacientes infectados con el virus del chikunguña. Con la ayuda de técnicas de análisis de redes complejas, inteligencia artificial y aprendizaje de máquinas, el grupo identificó la firma génica de la enfermedad, es decir, un conjunto de genes cuya expresión se altera debido a la interacción con el patógeno. Y luego mapearon el papel que cumplen en las células los genes implicados, como así también su importancia en el combate contra el virus.
Esta investigación, que se llevó a cabo en Brasil, contó con el apoyo de la FAPESP y estuvo coordinada por Helder Nakaya, docente de la Facultad de Ciencias Farmacéuticas (FCF) de la Universidad de São Paulo (USP). Participaron colaboradores del Instituto de Ciencias Biomédicas (ICB) de la USP, de la Facultad de Medicina de Ribeirão Preto (FMRP-USP), del Instituto Butantan y del Laboratorio Central de Salud Pública del Estado de Sergipe, entre otros científicos asociados.
“Identificamos también un conjunto de genes capaz de indicar, aún durante la fase aguda, si el paciente tiende a evolucionar hacia un cuadro de artralgia crónica [la inflamación de las articulaciones], relativamente común entre los infectados por el chikunguña. Sin embargo, este hallazgo aún deberá pasar por futuros estudios realizados con una cantidad mayor de muestras, a los efectos de que pueda confirmárselo”, declaró Nakaya a Agência FAPESP.
El artículo presenta los resultados de análisis realizados con muestras sanguíneas de 39 individuos nacidos en el estado brasileño de Sergipe que se infectaron durante la epidemia del año 2016. Esos datos se compararon con los de 20 personas del grupo de control, es decir, que no estaban infectadas y que vivían en la misma región que los pacientes estudiados.
El primer paso consistió en analizar el transcriptoma de las muestras, esto es, todas las moléculas de ARN mensajero (que codifican proteínas) y también los ARNs no codificantes (que no dan origen a proteínas, pero que desempeñan una acción reguladora en el genoma) expresados en las células que componen la sangre: hematíes, leucocitos y plaquetas. Al cuantificar los transcritos en las muestras, los investigadores pudieron medir el nivel de actividad de 20 mil genes y evaluar en cuáles de ellos la expresión aumentaba o disminuía durante la infección en comparación con lo que sucedía entre los integrantes del grupo de control.
“Nos enfocamos en los genes codificantes de proteínas [aquellos en los cuales se expresan los ARNs mensajeros], pues cumplen un papel cuya interpretación resulta más fácil. Es relativamente sencillo saber si estos codifican un receptor celular o un factor de transcripción, por ejemplo. De este modo, logramos entender mejor la patogénesis del chikunguña, es decir, de qué modo afecta el virus a las células y qué sistemas de defensa se activan en calidad de respuesta”, comentó Nakaya.
Este análisis reveló el mecanismo mediante el cual las células inmunitarias desencadenan el proceso inflamatorio para eliminar al virus, por ejemplo. Al conjunto de proteínas encargado de estructurar esta respuesta de defensa se lo conoce generalmente con el nombre de inflamasoma. Se trata de una maquinaria celular que responde al mando de distintas proteínas y produce diferentes moléculas proinflamatorias. En el caso de la infección ocasionada por el chikunguña, se observó que la mediación está a cargo de la enzima caspasa-1.
Este hallazgo se validó en el marco de experimentos con ratones realizados en colaboración con el investigador Dario Zamboni, de la FMRP-USP. Los investigadores –ambos ligados al Centro de Investigación en Enfermedades Inflamatorias (CRID), un Centro de Investigación, Innovación y Difusión (CEPID) de la FAPESP con sede en la USP de la localidad de Ribeirão Preto– observaron que en animales genéticamente modificados para no expresar la enzima caspasa-1, la infección por el chikunguña no induce la liberación de la molécula proinflamatoria denominada interleucina-1-beta (IL-1β), a diferencia de lo que sucede en los animales sin la referida alteración genética.
Similares, empero distintas
Tras identificar la firma génica de la infección por chikunguña –que abarca miles de genes cuya expresión se encuentra alterada en la enfermedad–, el grupo comparó los resultados con los obtenidos en muestras de pacientes infectados con el virus del dengue.
“Notamos que ambas firmas guardan una cierta similitud, pero algunos genes son específicos para el chikunguña. Y son esos los genes que podrán explotarse en estudios orientados hacia el desarrollo de fármacos”, dijo Nakaya.
En otro análisis, los investigadores compararon el perfil de expresión génica de los infectados por el chikunguña con el de personas que padecen artritis reumatoide, una enfermedad autoinmune caracterizada por la inflamación crónica en las articulaciones.
“En este caso, el objetivo consistía en descubrir la diferencia entre la artritis provocada por el virus y la artritis autoinmune, e identificar cuáles eran los genes específicos de la infección viral”, comentó Nakaya.
El análisis combinado de las tres firmas génicas reveló la existencia de 949 genes implicados únicamente en la artritis reumatoide, 632 tan solo en el dengue y 302 exclusivos del chikunguña. Siete genes aparecieron en las tres patologías simultáneamente: OAS1, C1QB, ANKRD22, IRF7, CXCL10, IFI6 y IFIT3.
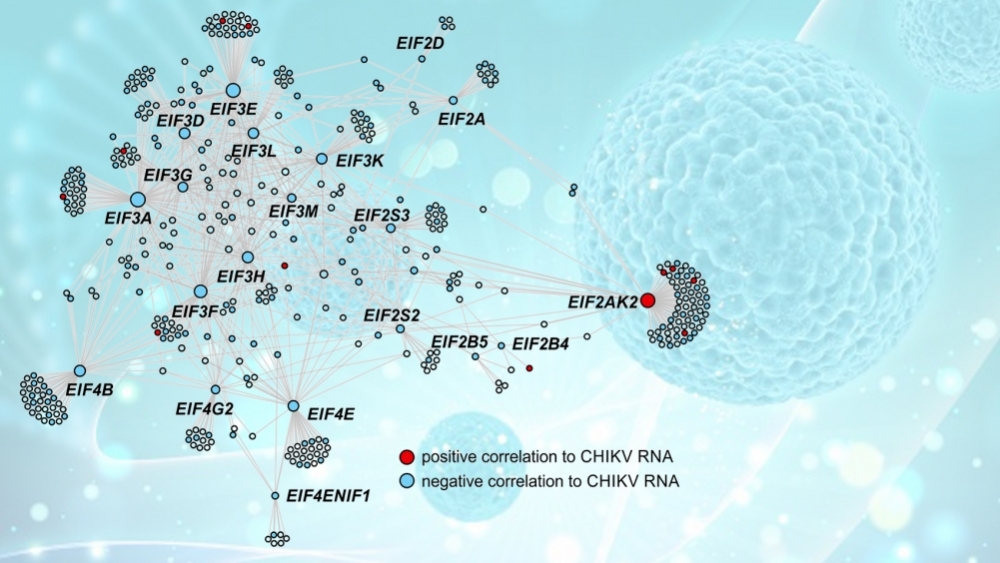
Con la ayuda de una herramienta llamada CEMiTool, desarrollada por Nakaya con el apoyo de la FAPESP, se efectuó un análisis de coexpresión para entender de qué manera interactúan los genes entre sí dentro de la red compleja existente en cada célula, formando vías de señalización y vías metabólicas.
“Esto nos permitió identificar ocho módulos principales [los módulos hacen referencia a genes con perfil similar de respuesta] e identificar en esas redes cuáles son los hubs, o nodos, es decir, aquellos genes con una mayor cantidad de conexiones y que, por tal motivo, son los más prometedores blancos para su explotación en la búsqueda de fármacos”, explicó.
El investigador remarcó que todos los datos de la investigación, tanto los brutos como los obtenidos mediante análisis, se encuentran disponibles en un repositorio público, y pueden consultarlos todos los interesados. También quedaron disponibles los códigos de programación que se emplearon en el artículo, para que otros investigadores puedan reproducir los resultados.
“Nuestro trabajo permitió generar una lista de potenciales dianas terapéuticas, y ahora estamos cruzando esos hallazgos con un banco de datos de compuestos activos. Ese cruzamiento se lleva a cabo en un medio computacional, pero con base en datos experimentales. Nos orientamos de acuerdo con estudios ya publicados que revelaron la existencia de drogas capaces de interferir en esos genes de interés”, dijo.
El grupo también sigue adelante con el análisis de los transcritos hallados en las muestras de los 39 pacientes infectados con chikunguña, ahora enfocándose en los ARNs no codificantes.
Nakaya contó con el apoyo de la FAPESP mediante una Ayuda a la Investigación – Regular y un Apoyo a Jóvenes Investigadores. Esta investigación también contó con apoyo en el marco de dos CEPIDs: el Centro de Investigación en Toxinas, Respuesta Inmune y Señalización Celular (CeTICS) y el CRID.
Puede leerse el artículo intitulado Systems analysis of subjects acutely infected with the Chikungunya virus, de Alessandra Soares-Schanoski et al. en el siguiente enlace: journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1007880.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
