Último episódio da série de reportagens faz um balanço sobre a viagem de um grupo de pesquisadores do Museu de Zoologia da USP que, ao longo de duas semanas, percorreu os rios Negro, Preto e Jauaperi, nos Estados do Amazonas e de Roraima
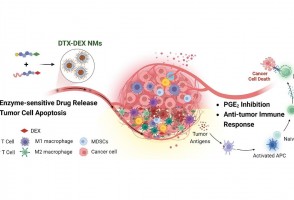
Em testes com animais, nanopartículas contendo substâncias já aprovadas para uso humano reduziram a inflamação no microambiente biológico em que cânceres desse tipo se instalam e vicejam, facilitando a ação do sistema imune
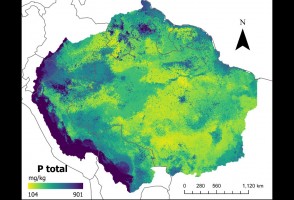
Pesquisa traz nova metodologia para descrever quantidade do mineral, que tem importância no ciclo de crescimento da vegetação e pode afetar a resposta da floresta às mudanças climáticas

Convidado é o professor da Universidade de Chicago Antonio Bianco, cuja pesquisa enfoca a ação dos hormônios tireoidianos
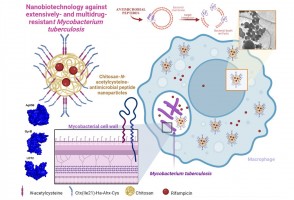
Pesquisadores da Unesp testaram a ação de partículas carregadas de antibióticos e outros compostos antimicrobianos em células infectadas pela bactéria causadora da doença. Resultados indicam que a estratégia é capaz de reverter o quadro de resistência aos medicamentos
Projeto conduzido por pesquisadores da USP e da Universidade de Birmingham foi um dos 15 apresentados em evento que celebrou a parceria entre a instituição britânica e a FAPESP. Ocasião também marcou o lançamento do University of Birmingham Brazil Institute, cuja missão é estreitar os laços de pesquisa com o Brasil
FAPESP lança edital em parceria com a M-ERA.NET, rede financiada pela União Europeia; pré-propostas serão recebidas até 14 de maio
CEPID B3/IQ-USP
Inscrições até 30/04/2024
IFSC-USP
Inscrições até 30/04/2024
SemeAr/Embrapa Agricultura Digital
Inscrições até 30/04/2024
FFCLRP-USP
Inscrições até 30/04/2024
IF-USP
Inscrições até 30/04/2024








Uma das artes de pesca utilizadas para coletar peixes-elétricos na Expedição DEGy Rio Negro foi empregada pela primeira vez em larga escala em água doce no projeto Calhamazon, que reuniu pesquisadores do Brasil e dos Estados Unidos entre 1993 e 1996

Baixa gravidade da lesão, atendimento médico rápido e cuidados adequados fizeram com que pesquisador pudesse voltar aos trabalhos no mesmo dia em que foi ferroado por peixe peçonhento. Na Amazônia, casos muitas vezes se agravam por carência de assistência especializada

Os mais abundantes peixes-elétricos estão presentes desde o fundo dos grandes rios até os igarapés, onde podem se enterrar na areia ou se confundir com o folhiço. Em duas semanas, expedição na bacia do rio Negro coletou 27 espécies do grupo
02/03/2024 a 30/04/2024
30/03/2024 a 27/04/2024
06/04/2024 a 08/06/2024
09/04/2024 a 30/04/2024
22/04/2024 a 26/04/2024
24/04/2024 a 24/04/2024

Startup apoiada pelo PIPE-FAPESP está desenvolvendo helicóptero autônomo para pulverizar plantações em terreno íngreme

Startup apoiada pelo PIPE-FAPESP está desenvolvendo helicóptero autônomo para pulverizar plantações em terreno íngreme
Comunicar Ciência
Prazo: 22/01
Belmont Forum Climate, Environment, and Health
Prazo: Jan 2024
PIPE Start FAPESP-Sebrae: iniciando a jornada empreendedora de base tecnológica
Prazo: 18/03
Centros de Pesquisa em Inteligência Artificial Aplicada à Saúde
Prazo: 18/03
Fundação Nacional de Ciência da Suíça
Prazo: 22/03
Apoio a pesquisa em citricultura
Prazo: 31/03
Expedições Científicas Amazônia+10
Prazo: 29/04
CEPID B3/IQ-USP
Inscrições até 30/04/2024
IFSC-USP
Inscrições até 30/04/2024
SemeAr/Embrapa Agricultura Digital
Inscrições até 30/04/2024
FFCLRP-USP
Inscrições até 30/04/2024
IF-USP
Inscrições até 30/04/2024
CEPID B3/IQ-USP
Incrições até 30/04/2024
IFSC-USP
Incrições até 30/04/2024
SemeAr/Embrapa Agricultura Digital
Incrições até 30/04/2024
FFCLRP-USP
Incrições até 30/04/2024
IF-USP
Incrições até 30/04/2024